Clustering of Time Course GeneExpression Data via Mixture









 clustering is given in terms of the estimated posterior probabilities of")
 Wolfe (NORMIX, 1965, 1967, 1970) It was the")
 Wolfe (NORMIX, 1965, 1967, 1970) It was the")
 • Titterington, Smith, and Makov (1985)")
 • Titterington, Smith, and Makov (1985) • Mc.")




/2 parameters. Σi = σ2 Ip")
 introduced a parameterization of the componentcovariance matrix Σi based on")





 data, it is assumed")

")

, with")
. Mixtures of linear mixed models for clustering gene")
 n=237 genes at p=17 time")



 0, 10, 20, …, 160 T is")











 synchronization where the yeast")



- Slides: 54
Clustering of Time Course Gene-Expression Data via Mixture Regression Models Geoff Mc. Lachlan (joint with Angus Ng and Sam Wang) Department of Mathematics & Institute for Molecular Bioscience University of Queensland ARC Centre of Excellence in Bioinformatics http: //www. maths. uq. edu. au/~gjm
Institute for Molecular Bioscience, University of Queensland
Time-Course Data Time-course microarray experiments are being increasingly used to characterize dynamic biological processes. (Microarray technology provides the ability to measure the expression levels of thousands of genes at once. ) In these experiments, gene-expression levels are measured at different time points, possibly in different biological conditions (e. g. treatment-control). The focus here is on the analysis of gene-expression profiles consisting of short time series of log expression ratios for each of the genes represented on the microarrays.
CLUSTERING OF GENE PROFILES can provide new insight into the biological proces of interest (coexpressed genes can contribute to our understanding of the regulatory network of gene expression). can also assist in assigning functions to genes that have not yet been functionally annotated. a secondary concern is the need for imputation of missing data
The biological rationale underlying the clustering of microarray data is the fact that many coexpressed genes are coregulated. It becomes a way of identifying sets of genes that are putatively coregulated, thereby generating testable hypotheses; see Boutros and Okey (2005). It assists with: the functional annotation of uncharacterised genes the identification of transcription factor binding sites the elucidation of complete biological pathways
Outline of Talk 1. Mixture model-based approach to analysis of gene-expressions 2. Normal Mixtures 3. Modifications for high-dimensional and/or structured data 4. Mixtures of linear mixed models 5. Clustering of gene profiles
Finite Mixture Models • Provide an arbitrarily accurate estimate of the underlying density with g sufficiently large • Provide a probabilistic clustering of the data into g clusters - outright clustering by assigning a data point to the component to which it has the greatest posterior probability of belonging.
Definition We let Y 1, …. Yn denote a random sample of size n where Yj is a p-dimensional random vector with probability density function f (yj) where the f i(yj) are densities and the pi are nonnegative quantities that sum to one.
By Bayes Theorem, for i=1, …, g; j=1, …, n. The quantity ti(yj; Y (k)) is the posterior probability that the jth member of the sample with observed value yj belongs to the ith component of the mixture.
A soft (probabilistic) clustering is given in terms of the estimated posterior probabilities of component membership A hard (outright) clustering is given by assigning each yj to the component to which it has the highest posterior probability of belonging; that is, given by the where
Multivariate Mixture Models Day (Biometrika, 1969) Wolfe (NORMIX, 1965, 1967, 1970) It was the publication of the seminal paper of Dempster, Laird, and Rubin (1977) on the EM algorithm that greatly stimulated interest in the use of finite mixture distributions to model heterogeneous data.
Multivariate Mixture Models Day (Biometrika, 1969) Wolfe (NORMIX, 1965, 1967, 1970) It was the publication of the seminal paper of Dempster, Laird, and Rubin (1977) on the EM algorithm that greatly stimulated interest in the use of finite mixture distributions to model heterogeneous data. Ganesalingam and Mc. Lachlan (Biometrika, 1978)
• Everitt and Hand (2001) • Titterington, Smith, and Makov (1985)
• Everitt and Hand (2001) • Titterington, Smith, and Makov (1985) • Mc. Lachlan and Basford (1988) • Lindsay (1996) • Mc. Lachlan and Peel (2000) • Bohning (2000) • Fruhwirth-Schnatter (2006)
Normal Mixtures Suppose that the density of the random vector Yj has a g-component normal mixture form where Y is the vector containing the unknown parameters.
One attractive feature of adopting mixture models with elliptically symmetric components, such as the normal or t densities, is that the implied clustering is invariant under affine transformations of the data, i. e. , under operations relating to changes in location, scale, and rotation of the data. Thus the clustering process does not depend on irrelevant factors such as the units of measurement or the orientation of the clusters in space.
Microarray Data represented as N x M Matrix Sample 1 Sample 2 Expression Signature Gene 1 Gene 2 Expression Profile Gene N Sample M M columns (samples) ~ 102 N rows (genes) ~ 104
Clustering of Microarray Data Clustering of tissues on basis of genes: latter is a nonstandard problem in cluster analysis (n =M << p=N) Clustering of genes on basis of tissues: genes (observations) not independent and structure on the tissues (variables) (n=N >> p=M)
The component-covariance matrix Σi is highly parameterized with p(p+1)/2 parameters. Σi = σ2 Ip (equal spherical) Σi = σi 2 Ip (unequal spherical) Σi = D (equal diagonal) Σ i = Di (unequal diagonal) Σi = Σ (equal)
Banfield and Raftery (1993) introduced a parameterization of the componentcovariance matrix Σi based on a variant of the standard spectral decomposition of Σi (i=1, …, g).
However, if p is large relative to the sample size n, it may not be possible to use this decomposition to infer an appropriate model for the component-covariance matrices. Even if it is possible, the results may not be reliable due to potential problems with nearsingular estimates of the componentcovariance matrices when p is large relative to n.
Hence, in fitting normal mixture models to high-dimensional data, we should first consider • some form of dimension reduction and/or • some form of regularization
Mixture Software: EMMIX for UNIX Mc. Lachlan, Peel, Adams, and Basford http: //www. maths. uq. edu. au/~gjm/emmix. html
PROVIDES A MODEL-BASED APPROACH TO CLUSTERING Mc. Lachlan, Bean, and Peel, 2002, A Mixture Model-Based Approach to the Clustering of Microarray Expression Data, Bioinformatics 18, 413 -422 http: //www. bioinformatics. oupjournals. org/cgi/screenpdf/18/3/ 413. pdf
Microarray Data represented as N x M Matrix Sample 1 Sample 2 Expression Signature Gene 1 Gene 2 Expression Profile Gene N Sample M M columns (samples) ~ 102 N rows (genes) ~ 104
In applying the normal mixture model to cluster multivariate (continuous) data, it is assumed as in most typical cluster analyses using any other method that (a) there are no replications on any particular entity specifically identified as such; (b) all the observations on the entities are independent of one another
For example, where and where
Clustering of gene expression profiles • Longitudinal (with or without replication, for example time-course) • Cross-sectional data EMMIX-WIRE EM-based MIXture analysis With Random Effects Ng, Mc. Lachlan, Wang, Ben-Tovim Jones, and Ng (2006, Bioinformatics) Supplementary information : http: //www. maths. uq. edu. au/~gjm/bioinf 0602_supp. pdf
In the ith component of the mixture, the profile vector yj for the jth gene follows the model
N(mi, Bi), with
• Celeux et al. (2005). Mixtures of linear mixed models for clustering gene expression profiles from repeated microarray measurements. Statistical Modelling 5 , 243 -267. • Qin and Self (2006). The clustering of regression models method with applications in gene expression data. Biometrics 62, 526 -533. • Booth et al. (2008). Clustering using objective functions and stochastic search. J R Statist Soc B 70, 119 -139.
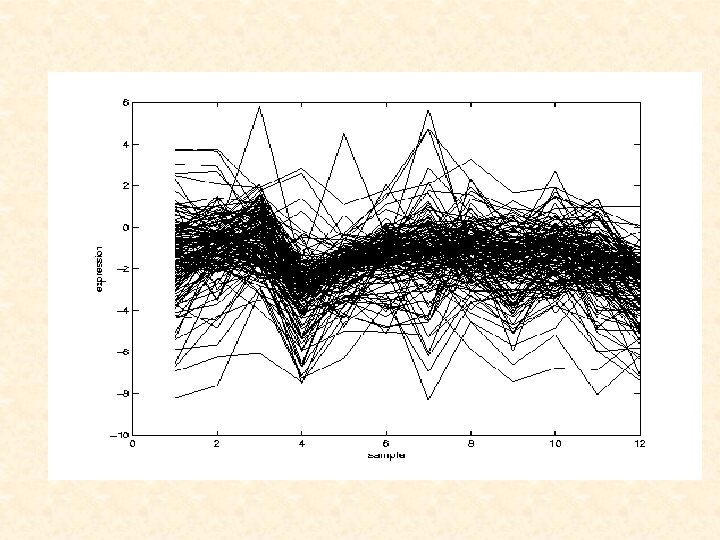
Yeast cell cycle data of Cho et al. (1998) n=237 genes at p=17 time points categorized into 4 MIPS (Munich Information Centre for Protein Sequences) functional groups. The yeast system is useful because of our ability to control and monitor the progression of cells through the cell cycle (temperaturebased synchronization with temperature-sensitive genes whose product is essential for cell-cycle progression).
High-density oligonucleotide arrays were used to quanitate m. RNA transcript levels in synchronized yeast cells at regular intervals (10 min) during the cell cycle (genes with cell-cycle dependent periodicity). Samples of yeast cultures were taken at 17 time points after their cell cycle phase had been synchronized. The data were reduced to a short time series of log expression ratios for each of the yeast genes represented on the microarrays (expression ratios were calculated by dividing each intensity measurement by the average for that gene.
Example. Clustering of yeast cell cycle time-course data n = 237 genes p = 17 time points where
In the ith cluster,
Estimated T following Booth et al. (2004) 0, 10, 20, …, 160 T is the period – estimated to be 73 min.
Table 1: Values of BIC for Various Levels of the Number of Components g The Number Of Components 2 3 4 5 6 7 10883 10848 10837 10865 10890 10918
Cluster-specific random effects term
Table 2: Summary of Clustering Results for g = 4 Clusters Model Rand Index Adjusted Rand Index Error Rate 1 0. 7808 0. 5455 0. 2910 2 0. 7152 0. 4442 0. 3160 3 0. 7133 0. 3792 0. 4093 Wong 0. 7087 0. 3697 NA
• The use of the cluster-specific random effects terms ci leads to a clustering that corresponds more closely to the underlying functional groups than without their use.
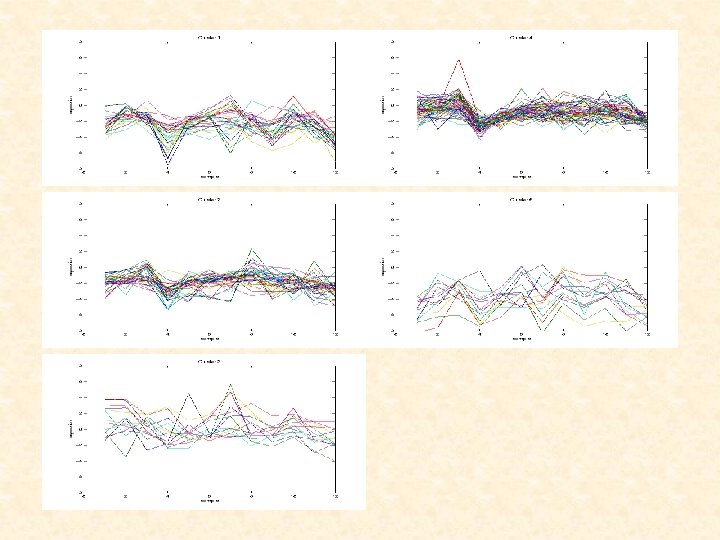
Figure 1: Clusters of gene-profiles obtained by mixture of linear mixed models with clusterspecific random effects
Figure 2: Clusters of gene-profiles obtained by mixture of linear mixed models without clusterspecific random effects
Figure 3: Clusters of gene-profiles obtained by mixtures of linear mixed models with and without cluster-specific random effects
Figure 4: Plots of gene profiles grouped according to their functional grouping
Figure 5: Plots of clustered gene profiles versus functional grouping
Figure 6: Clusters of gene-profiles obtained by k-means
Figure 7: Plots of Clusters of gene-profiles: Model-based clustering versus k-means
Another Yeast Cell Cycle Dataset Spellman (1998 used α-factor (pheromone) synchronization where the yeast cells were sampled at 7 minute intervals for 119 minutes; the period of the cell cycle was estimated using least squares to be T=53 min.
Example. Clustering of time-course data n = 612 genes p = 18 time points where
Clustering Results for Spellman Yeast Cell Cycle Data
Mixtures of linear mixed models Useful in modelling biological processes that exhibit periodicity at different temporal scales (not restricted to cell cycle data; e. g changes in core body temperature, heart rate, blood pressure). In summary, they provide a flexible tool to cluster highdimensional data (which may be correlated and structured) for a wide range of experimental designs, e. g. - longitudinal data (with or without replication) - cross sectional data (multiple samples at one time point). Provide an integrated framework for the analysis of microarray data by incorporating experimental design information and (biological or clinical) covariates.