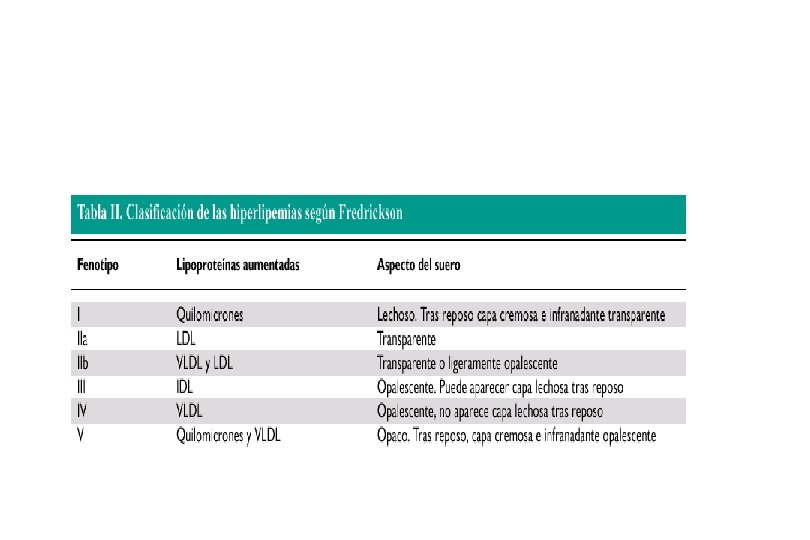
CLASIFICACION DE FREDRICKSON FENOTIPO I quilom LPP LAB

CLASIFICACION DE FREDRICKSON FENOTIPO • I quilom. LPP LAB TG +++ ? ATEROG. Quilom. Fam (FLPL def. apo C-II def). • IIa • LDL C ++ +++ FH, FCH, HCP, defecto apo B • • II b LDL y VLDL C/Tg ++ +++ FH, FCH DLP 2º Disglobulinemia, pancreatitis, DBT mal controlada. Hipotiroidismo, PIA, S. Nefrótico, anorexia nerviosa. Hipotiroidismo, PIA, S. Nefró tico, anorexia nerviosa. • • • III IV IDL VLDL C ++ TG ++/C N/+ +++ + Disbetalipoprot. Fam. FTG DBT, hipotiroidismo, disglob. Enf por depósito de gluc, hipotir, LES, DBT, IR, etanol, S nefrótico. • • • DLP 1º V VLDL - quil. TG +++ CN/+ Manual of Lipid Disorders. Gotto A. 2 nd edition. 1999. + HTGCF DBT, enf. por depósito d glucógeno, hipotiroidismo, S. Nefrótico, embarazo, disglobulinemia.

HIPERLIPOPROTEINAMIAS 1 - PRIMARIAS. 2 - SECUNDARIAS.
 HIPERCOLESTEROLEMIA : • fenotipo IIa : HF –")
HIPERLIPOPROTEINEMIAS COLESTEROL y/o TRIGLICERIDOS • 1) HIPERCOLESTEROLEMIA : • fenotipo IIa : HF – defecto Apo B-100 (BDF). • Hipercol poligénica-Hip Comb Fliar • 2) HIPERTRIGLICERIDEMIA : • fenotipo I, IV o V (con/sin Q) : HTGF-Htg poligénica • Hip Comb Fliar • 3) MIXTAS : • fenotipo IIb o III : HCF, DBLF.

HIPERLIPOPROTEINEMIAS FAMILIARES • 5% de la población con un transtorno heredado de las LPP. • 1) aumento de colesterolemia y/ trigliceridemia y de las LPP. • 2) mayor riesgo de ECV (no en la hiperquilomicronemia fam). • 3) depósitos extravasculares (piel, tendones, hígado).

DISLIPOPROTEINEMIAS • HIPERCOLESTEROLEMIA • HIPERTRIGLICERIDEMIA Primarias • HIPERLIPEMIA MIXTA • DISMINUCION DE HDL Secundarias

DISLIPOPROTEINEMIAS Monogénicas Poco frecuentes con alto impacto Hipercolesterolemia familiar Hiperquilomicronemia Variantes de DNA comunes • Bajo impacto Poligénicas Frecuentes Hipercolesterolemias e Hipertrigliceridemias Variantes de DNA poco comunes • Alto impacto

FENOTIPOS LIPIDICO-LIPOPROTEICO ASOCIADOS A RIESGO CARDIOVASCULAR ¨Hipercolesterolemias ¨Hiperlipemias mixtas ¨Hipoalfalipoproteinemias

Hipercolesterolemia: Hipercolesterolemia severa: todo sujeto con valores de colesterol-LDL> 190 mg/dl (independientemente de la causa) • Hipercolesterolemia Autosómica Dominante (ADH): pacientes con mutaciones en genes que controlan los niveles de LDL (FH, APOB, PCSK 9) • Hipercolesterolemia Familiar (FH): desorden monogénico que afecta al R-LDL Ho. FH He. FH ADH: FH 95% Apo. B (4 -5%) PCSK 9 (<1%)
 • Defecto familiar")
Desórdenes monogénicos y genes relacionados con hipercolesterolemia • Hipercolesterolemia familiar (LDLR) • Defecto familiar de apo. B 100 (apo. B) • Hipercolesterolemia autosómica (PCSK 9) • Deficiencia de colesterol 7 alfa-hidroxilasa (CYP 7 AI) • Sitosterolemia familiar (ABCG 5 y ABCG 8) • Hipercolesterolemia asociada a apo E (apo. E)

HIPERCOLESTEROLEMIAS PRIMARIAS Aumento col-LDL ¨Hipercolesterolemia Familiar ¨Defecto familiar de apo B 100 ¨Otras formas (ARH, PCSK 9) ¨Hipercolesterolemia poligénica

Hipercolesterolemia Familiar 1. HMG Co. A Reductasa Receptor LDL Nu CL 2. ACAT CE LDL Colesterol ester hidrolasa; p. H=7 CE= FA+CL 3. Receptor LDL Colesterol ester hidrolasa; p. H=4 FA + CL AMINOACIDOS HDL LCAT C Apo. A 1
 < 2% actividad normal DEFECTO-RECEPTOR 2 – 25")
RELACION C - LDLr RECEPTOR (-) < 2% actividad normal DEFECTO-RECEPTOR 2 – 25 % actividad norm.

HIPERCOLESTEROLEMIA FAMILIAR * Descripta por Fagge en 1873. * Causa : mutaciones en el gen del r-LDL (cromosoma 19). 50 % de heterocigotas con mutaciones en el gen r-LDL. * Transmisión autosómica dominante con penetración completa. * HCF heterocigota : 1 cada 500 individuos (1 c/80 en sudafricanos descendientes de franceses o alemanes, en libaneses y francocanadienses). * HCF homocigota : 1 cada 1. 000 individuos. * hipercolesterolemia desde el nacimiento (heterocigotas N x 2; 350 mg%, homocigotas Nx 6 -8; 900 -1000 mg%). * tiempo de circulación de las LDL prolongado. * HDL generalmente bajo(homocigotas). * TG generalmente normales (altos : mal pronóstico-obesidad). * Fenotipo IIa (II b raro). • * Lp(a) aumentada. • * a veces aumento de la absorción del colesterol (co-herencia con el geno • tipo epsilon A de las apo A). • • •

HCF : clínica • 1 - XANTOMAS : depósitos de células en espumadera. • * xantomas tendinosos (tendón de Aquiles) y en nu • dillos de las manos. • Diagnósticos diferenciales : • * xantomatosis cerebro tendinosa. • * fitoesterolemia. • 2 - arco senil-xantelasmas precoces (inespecíficos).

Características de la Hipercolesterolemia Familiar ¨ Hipercolesterolemia desde el nacimiento ¨ Xantomas tendinoso y cutáneos, arco corneal, xantelasmas, engrosamiento del tendón de Aquiles ¨ Aterosclerosis prematura, cardiopatía isquémica precoz ¨ Herencia autosómica dominante ¨Prevalencia: -1/500 Heterocigotas Franco-Canadienses (1/270), Libaneses (1/170), Sudafricanos blancos(1/100) - 1/1 millón Homocigotas Xantomas planos Arco corneal Xantelasmas

XANTOMAS – ARCO SENIL

C : 550 mg%. TG : 180 mg%. C-HDL : 34 mg%. C-LDL : 480 mg% N Engl J Med 2005; 352 : 2424
 Soutar A, Naoumova P, Nature 2007; 4:")
Mutaciones del gen LDL-R (más de 1700) Soutar A, Naoumova P, Nature 2007; 4: 4

HIPERCOLESTEROLEMIAS PRIMARIAS Aumento col-LDL ¨Hipercolesterolemia Familiar ¨Defecto familiar de apo B 100 ¨Otras formas (ARH, PCSK 9) ¨Hipercolesterolemia poligénica
 R-LDL apo. B ¨ 2(p 24 -p 23)")
Defecto Familiar de apo. B-100 (FDB) R-LDL apo. B ¨ 2(p 24 -p 23) - 43 Kb (29 ex. ) - 4536 aminoácidos ¨FDB: Arg 3500 Gln - CGG CAG - ex. 26 ¨Herencia autosómica dominante ¨Prevalencia 1/1000 (N Engl J Med 1998) ¨Otras mutaciones: Arg 3531 Cys; Arg 3500 Trp

HIPERCOLESTEROLEMIAS PRIMARIAS Aumento col-LDL ¨Hipercolesterolemia Familiar ¨Defecto familiar de apo B 100 ¨Otras formas menores (PCSK 9, ARH) ¨Hipercolesterolemia poligénica
 Mutaciones en el gen")
Mutación de la Proteína convertasa subtilisin/kexin tipo 9 (PCSK 9) Mutaciones en el gen codificante cosegrega con severa hipercolesterolemia q El mecanismo aun se discute, pero PCSK 9 es un gen regulado por esteroles q La sobre-expresión reduce el nivel de receptores-LDL q Soutar A, Naoumova P, Nature 2007; 4: 4 Soutar A, Curr Opinion Lipidol 2011, 22: 192

Papel de la PCSK 9 sobre el receptor-LDL La mutación de PCSK 9 asociada a hipercolesterolemia, resulta en una proteína que se une 10 veces más fuerte al receptor y promueve una degradación aún mayor.
 ü Herencia recesiva ü LDL-R normal pero falla en internalizar")
Hipercolesterolemia Autosómica Recesiva (ARH) ü Herencia recesiva ü LDL-R normal pero falla en internalizar LDL ü Mutación del gen LDL receptor adaptor protein 1 (LDLRAP 1 ó ARH) ü El fenotipo es semejante al de pacientes con defecto del LDL-R, pero con niveles menores de colesterol total y colesterol-LDL (mayor colesterol-HDL) ü Muestran mejor respuesta al tratamiento farmacológico
 col-LDL>500 mg/dl 1/1. 000")
ADH • Ho. FH: R-LDL marcadamente disfuncional (2 -30% actividad) col-LDL>500 mg/dl 1/1. 000 • He. FH: R-LDL disfuncional (50% actividad) col-LDL (2 -3 veces valor promedio) 1/200 -500
 q ECV")
DIAGNOSTICO DE HIPERCOLESTEROLEMIA FAMILIAR q HISTORIA FAMILIAR DE HIPERCOLESTEROLEMIA (especialmente en niños) q ECV PREMATURA q NIVEL DE COLESTEROL TOTAL Ho. FH>400 mg/dl; He. FH>190 mg/dl adultos y >160 mg/dl niños q XANTOMAS q ARCO CORNEAL (en menores de 45 años)

Distintos sets diagnósticos de HF utilizados en la clínica MEDPED Concentración de colesterol-LDL (valores de corte que proveen 98% de especificidad) Historia Familiar q Scientific Steering Commitee on Behalf of the Simon Broome Register Group. Valores lipídicos pre-tratamiento Xantomas tendinosos Historia familiar de infarto prematuro de miocardio y/o hipercolesterolemia q Dutch Lipid Clinic Network Valores de col-LDL pre-tratamiento Otras manifestaciones clínicas Historia familiar q


Hipercolesterolemia Familiar es un problema mundial q Diez millones de personas afectadas q 200. 000 pacientes morirán prematuramente por ECV q 80 % de los FH permanecen sin diagnóstico q 84 % de los FH permanecen sin tratamiento farmacológico q <10% alcanzan el objetivo de col-LDL

Búsqueda de casos índices y Screening en cascada: Potenciales casos índices deberían buscarse entre: – pacientes menores de 60 años con ECV, así como entre aquellos que concurran a programas de rehabilitación cardíaca – Pacientes con xantomas tendinosos y arco corneal prematuro El laboratorio debe alertar ante pacientes con col-LDL>190 mg/dl ó col-total>270 mg/dl El diagnóstico deberá realizarse con dos medidas de col-LDL en ayunas y alguno de los algoritmos propuestos Niños con probable FH deberían estudiarse antes de los 10 años (con historia familiar en primer grado antes de los 55 años) utilizando estrategias de screening en cascada, universal o selectiva. Screening buscando genotipos o fenotipos. Si el estudio del DNA no es posible se evaluará edad, sexo y valores de col-LDL El screening en cascada debería comenzar con familiares en primer grado y luego extenderse a familiares en segundo y tercer grado.

LDL AFERESIS Indicaciones de la LDL-aféresis • 1. HF homocigota, con colesterol LDL > 500 mg/dl • 2. HF heterocigota, con colesterol LDL > 300 mg/dl • 3. HF heterocigota con enfermedad coronaria y colesterol LDL > 200 mg/dl Cifras con tratamiento farmacológico
 Lecho")
LDL-aféresis : Unión selectiva de lipoproteínas con apo B LDL, VLDL, IDL, LP(a) Lecho de Dextran sulfato celulosa Reduce LDL-C ~80% Se utiliza cada 2 semanas FDA lo aprovó en 1997 Transplante Hepático: ~70% de los R-LDL se encuentran en hígado Alto riesgo, período prolongado de inmuno-supresion, alto costo

HIPERCOLESTEROLEMIAS PRIMARIAS Aumento col-LDL ¨Hipercolesterolemia Familiar ¨Defecto familiar de apo B 100 ¨Otras formas menores (ARH, PCSK 9) ¨Hipercolesterolemia poligénica

Hipercolesterolemia Poligénica q Frecuencia: > 50 % Hipercolesterolemia Primaria q Edad de manifestación >20 años q Aterosclerosis prematura. Xantomas poco frecuentes q Factores ambientales q Gen/es: cuál/es ? modo de herencia ?

HIPERCOLESTEROLEMIA POLIGÉNICA: Individuos con col-LDL>P 95 para su edad y sexo, en los cuales se haya descartado una dislipemia secundaria ó alguna de las otras hipercolesterolemias primarias.
- Slides: 36