CHILDHOOD POISONIONG PARACETAMOL POISONING Acetaminophenparacetamol is a remarkably

CHILDHOOD POISONIONG

• PARACETAMOL POISONING:

• Acetaminophen/paracetamol is a remarkably safe and effective drug at appropriate dosages. • Following oral ingestion, PCT is rapidly absorbed with a time to peak of 45 minutes. Liquid acetaminophen has a time to peak of 30 minutes. • In delayed release tablets, peak PCT concentrations may be delayed 8 hours or longer. • First-pass metabolism removes 25% of a therapeutic dose. Once absorbed, approximately 90% of acetaminophen normally undergoes hepatic conjugation with glucuronide and sulfate to form inactive metabolites, which are eliminated in the urine

• The remaining fraction is oxidized by enzymes of the cytochrome system resulting in the formation of N-acetyl-pbenzoquinoneimine (NAPQI). • Glutathione (GSH) quickly combines with NAPQI, and the resulting complex is converted to nontoxic conjugates, which are eliminated in the urine. • The elimination half-life of PCT is approximately 2 -3 h after a nontoxic dose but may become prolonged in patients with liver disease.

Toxicokinetics: • The dose associated with toxicity are >150 mg/kg/d. • After an acute overdose, peak plasma concentrations generally occur within 4 hours. • Increased risk for hepatic toxicity: Acute febrile illness, poor nutritional status, chronic use of CYP 450 enzyme-inducing drugs such as rifampin or phenobarbital • Toxic metabolites accumulate further as elimination becomes prolonged when normal metabolic systems become saturated.

Pathophysiology: • After therapeutic PCT dosing, GSH supply is in excess of that required to detoxify NAPQI, and no toxicity occurs. • After overdose, the rate and quantity of NAPQI formation exceed GSH supply and regeneration, resulting in free NAPQI. • Free NAPQI rapidly binds to hepatocyte constituents and causes hepatic injury.

Clinical course: • Stage I: o Nonspecific clinical findings include nausea, vomiting, malaise, pallor and diaphoresis. o Rarely in massive overdose, a decreased level of consciousness and metabolic acidosis may occur (even in the absence of hepatotoxicity). o Liver function tests are normal. • Stage II: o Represents the onset of liver injury o Onset: within 24 -36 hours after ingestion. o Symptoms and signs mimic other causes of hepatocellular injury. o SGOT abnormalities occur before ↑PT/INR, elevated bilirubin, hypoglycemia and metabolic acidosis.

• Stage III: o This is the time of maximal hepatotoxicity and occurs between 72 and 96 hr after ingestion. o Fulminant hepatic failure (FHF) with encephalopathy or severe coagulopathy may occur. o SGOT and SGPT >10, 000 IU/L are common. Abnormalities of PT, bilirubin, glucose, lactate and p. H indicate worse severity. o Fatalities from FHF generally occur between 3 and 5 days after an acute overdose. o Death results from complications of multiorgan failure, including hemorrhage, ARDS, sepsis and cerebral edema.

Stage IV: Recovery phase Hepatic regeneration occurs, which is complete in survivors. In most cases, LFT is normal by 5 -7 days after an acute overdose. o Recovery may take longer in severely poisoned patients • o o o

• o o o Other organ injury: Renal insufficiency Myocardial injury Pancreatitis

Diagnostic evaluation: • The Rumack Matthew nomogram o When using the PCT nomogram, it is essential to precisely define the time window during which exposure occurred and, if the time is unknown, to use the earliest likely time as the time of ingestion. o Using this approach, patients with peak levels below the treatment line do not require further evaluation or treatment for acute acetaminophen overdose.
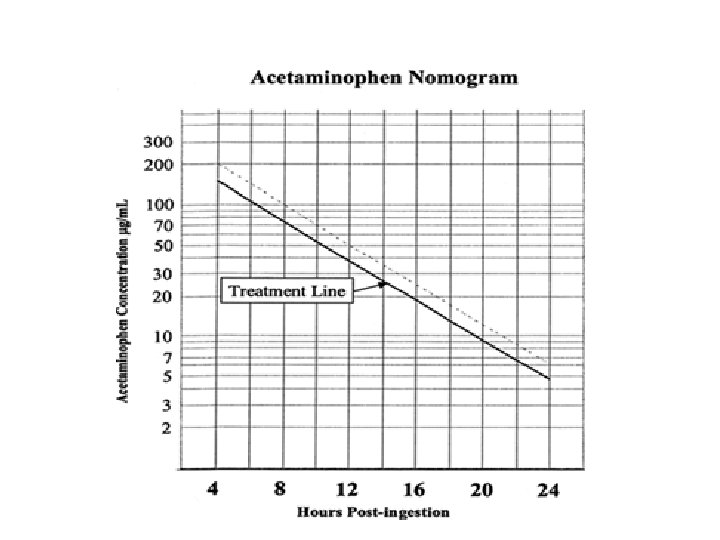

• The level may be plotted on the nomogram and NAC either continued or discontinued based on the result. • If levels or results of levels can’t be obtained within 8 hours, the history OR an ingestion that has the potential to cause toxicity is sufficient to initiate and complete a course of NAC therapy.

• Situations in which the Rumack- Matthew nomogram is of limited use: – Serum PCT levels obtained prior to 4 hours post-ingestion are not interpretable because of ongoing drug absorption and distribution – In chronic ingestion or in overdose with an extended-release preparation, the nomogram is less predictive of toxicity. – The nomogram is unreliable for high-risk populations (see above). – Accurate assessment on nomogram requires a precise of ingestion, which may not be available. – Patients who present after 24 hours • In such cases, if SGOT is elevated OR PCT levels elevated, treat with NAC

Management: • Limiting GI Absorption: – Administration of activated charcoal shortly after ingestion decreases the peak levels. – Most effective when given within the first 1 -2 hours, can be upto 4 hours post ingestion. • Supportive Care: – Control nausea and vomiting – Managing the hepatic failure as per protocol – Management of renal dysfunction. – Monitor and treat hypoglycemia. – Administration of FFP/blood products should be based on presence of bleeding rather than PT/INR alone.
: – NAC is most effective if administered within the first")
N- Acetyl Cysteine (NAC): – NAC is most effective if administered within the first 8 -24 hr of ingestion. If time of ingestion is > 24 hr, NAC is indicated if hepatotoxicity is present. – Three distinct roles of NAC • Limits the formation of NAPQI • Increases GSH availability • Nonspecific antioxidant effects preserves multi-organ function – NAC dose: Intravenous regimen: Use 20% NAC (200 mg/m. L) as infusions. – – Dilute 20% NAC solution to 3% solution with 5% dextrose. 150 mg/kg over 60 min 50 mg/kg over next 4 hours 100 mg/kg over next 16 hrs
 and dilute")
– NAC dose: Oral regimen • Use 10% NAC (100 mg/m. L) and dilute 2: 1 in water or juice to make a 5% solution (50 mg/m. L). • Initial dose: 140 mg/kg • Maintenance dosage: 70 mg/kg every 4 h for 17 doses – Intravenous and oral NAC are equally efficacious in treating acetaminophen toxicity. Oral absorption may take some time to occur in comparison to an IV dose. Vomiting may decrease the oral dose retained and also increases the risk of aspiration.

– Intravenous NAC regimen is indicated for the following: oral NAC not tolerated despite adequate antiemetic therapy, GI bleeding or obstruction, neonatal acetaminophen toxicity from materbal overdose. – Side effects of NAC are nausea, vomiting, diarrhea, skin rash, anaphylaxis and hyponatremic seizures. – NAC should be continued for at least 72 hours. Levels of PCT and SGOT concentration should be measured at the completion of the course, and NAC therapy should be continued beyond the prescribed time if there is evidence of significant liver injury (SGOT greater than normal) or acetaminophen metabolism is incomplete (levels >10 µg/m. L).

– For patients who develop liver failure, IV NAC is continued until PT is near normal and encephalopathy, if present, is resolved.

• King's College Hospital criteria for liver transplantation after PCT overdose: o A single criterion of p. H <7. 30 after fluid and hemodynamic resuscitation OR o A combination of 3 criteria, i. e. , PT >100 seconds, creatinine >3. 3 mg/d. L, and grade III/ IV encephalopathy is predictive of a patient who will die without transplant. o Any patient who meets or approaches these criteria should be considered for liver transplant.

• CORROSIVE POISONING

• Corrosives are compounds that cause tissue injury by chemical reactions. • Approximately 80% of caustic ingestions occur in children younger than 5 years. • Common acid-containing sources: Toilet bowl cleaning products, automotive battery liquid, rust removal products, metal, cement and drain cleaning products. • Common alkaline-containing sources: Ammonia-containing products, drain, oven and swimming pool cleaning products, dishwasher detergent, bleaches, button batteries.

Pathophysiology: • Alkalis: o Alkalis cause liquefaction necrosis, a process resulting from the saponification of fats, dissolution of proteins and emulsification of lipid membranes. o The resultant tissue sloughing permits the alkali to penetrate to deeper levels. o Alkali induced injury thus > acid induced injury o Tissue injury progresses rapidly over the first few minutes but can continue for several hours. Over the ensuing 4 days, bacterial infection and inflammation cause additional injury. Granulation tissue then develops, but collagen deposition may not begin until the second week. The tensile strength of healing tissue is lowest during the first 2 weeks. Epithelial repair may take weeks to months. Scar retraction begins in the third week and continues for months as collagen is laid down and undergoes maturation. o The most severely injured tissues are those that first contact the alkali, that is the squamous epithelial cells of the oropharynx, hypopharynx, and esophagus. The esophagus is the most commonly involved organ with the stomach much less frequently involved after alkaline ingestions.

Acids o Acid burns are characterized by coagulation necrosis. Protein is denatured, resulting in the formation of a firm coagulum or eschar with a white, plaque-like appearance. o This eschar may protect the underlying tissue from further damage. o The stomach is the most commonly involved organ following an acid ingestion. Small bowel exposure also occurs in about 20% of ingestions. o Significant exposures may also result in absorption of the acidic substances leading to systemic metabolic acidosis , hemolysis, acute renal failure, and death. o In general, solids tend to produce intense localized upper esophageal injury, while liquid acids tend to produce circumferential lesions in the distal esophagus. Areas of anatomical narrowing, such as at the cricopharynx or the carina are subject to longer contact time and may be associated with more severe injury.

Clinical Manifestations: • Eyes: o Chemical burns to the eye range from mild to severe (permanent vision loss). o Eye pain, blepharospasm, conjunctival hemorrhages, and chemosis are seen in all grades of injury. • Skin: o Differences exist between thermal and chemical burns of the skin. o Chemical burns rarely blister, and the affected skin is usually dark, insensate, and firmly attached regardless of the burn depth. o Healing usually takes longer than for thermal burns.

• Airway obstruction may occur secondary to edema, bleeding, and necrosis. o Potential for airway swelling or esophageal perforation exists, even if there are no o symptoms initially or no oropharyngeal burns o Signs of impending airway obstruction may include the following: Stridor, hoarseness, aphonia, respiratory distress, tachypnea, hyperpnea, cough, desaturation.

• GI tract: o Ingestion of caustic materials may produce oral burns with reddened areas or whitish plaques. o Symptoms include oropharyngeal pain, dysphagia with drooling and persistent salivation, chest or abdominal pain, vomiting, GI bleeds (blood or tissue). o Signs of perforation with mediastinitis include chest pain, respiratory distress, fever, subcutaneous emphysema of the chest or neck, pleural rub. o Fluid losses from vomiting, third spacing, and GI bleeding may lead to hypovolemic shock. o In patients who survive the initial phases of injury, late problems include strictures, fistula, hypomotility disorders and an increased risk of gastrointestinal cancers. o Hypocalcemic arrhythmias can develop precipitously after ingestion of significant amounts of hydrogen fluoride (found in some rust removers).

Management: • Stabilization of the ABCs takes priorities in any situation, then decontamination. o Symptomatic patients require special attention to the airway. o If airway obstruction present or imminent, early expert fiberoptic intubation (if available) preferred to standard orotracheal intubation. Intubation may further traumatize damaged areas or perforate the pharynx. Surgical airway may be required. o Assess circulatory status and initiate volume resuscitation as necessary

• Decontamination: • Eyes: o Early, copious, repeated irrigation must be performed as damage increases with prolonged contact with corrosives. Tap water or any liquid that is clear and drinkable can be used in the prehospital setting. Normal saline and lactated Ringer's solution are traditionally used irrigation fluids in hospital settings. o Irrigation should initially be performed for about 20 minutes. Prolonged irrigation (up to 24 hours) may be beneficial for strong alkalis. o Blepharospasm secondary to pain can prevent effective irrigation so instillation of a topical anesthetic is recommended.

• Skin: – Remove all contaminated clothing. – Prompt irrigation with copious amounts of water for at least 15 minutes for acid exposures and 30 minutes for alkali exposures. Longer irrigation is recommended for alkalis because they have detergent properties. – Patients with second- or third-degree skin burns should be referred to a surgeon. – Definitive management includes aggressive early debridement and grafting

• GI Tract: – Rinsing with water or saline is recommended for mouth exposures. – Emesis/ ipecac is contraindicated because of aspiration risk and increased severity of esophageal and laryngeal burns. – Gastric aspiration and lavage are contraindicated. – Activated charcoal is contraindicated because it does not bind these agents and also may create a problem with visualization during endoscopy. – Maintain NPO status. Attempts to neutralize the caustic substance are ineffective and obscure and delay endoscopy. – Neutralization with weak acids or bases is contraindicated as they may lead to the production of heat which may promote furthermal injury in addition to corrosive effects.
")
• General management: – Ensure that all patients take nothing per mouth (NPO) until the extent of injury has been determined. Patients can commence oral fluids when they are able to swallow their own secretions. – Obtain early surgical and GI consultation; early endoscopy usually indicated. – Passage of a nasogastric tube is hazardous because of perforation risk: however, some centres advocate careful insertion during scopy as it permits enteral nutrition and may also have a role in preventing stricture by stenting the esophagus. – Steroids are controversial but widely used. Useful in the presence of airway compromise or stridor. Dose: Methylprednisolone 0. 5 mg/kg/dose Q 6 H. – Prophylactic antibiotics have no role unless perforation has occured, however, monitor carefully for secondary infection. – Symptomatic relief can be provided with antacids, sucralfate, H 2 -blockers, analgesics. – Surgical exploration is indicated if perforation or penetration into surrounding tissues is suspected by findings such as fever, progressive abdominal or chest pain, hypotension, or signs of peritonitis or proved by endoscopic or radiographic findings. – Stricture formation is usually treated with endoscopic dilatation beginning 3 to 4 weeks after ingestion.

• Investigations: o Patients with signs and symptoms suggestive of significant injuries should have an ECG, blood gas analysis, complete blood cell count, type and cross-match, coagulation profile, electrolytes, glucose, and liver and renal function o A chest X-ray is indicated in patients with signs of a significant ingestion or respiratory distress. Patients with abdominal pain or tenderness require an abdominal X-ray to exclude the presence of free air, indicating perforation. o CXR: Findings may include esophageal perforation, mediastinitis, pleural effusions, pneumoperitoneum, aspiration pneumonitis, or a button battery. However, the absence of findings does not preclude perforation or other significant injury. o Abdominal radiography: Findings may include pneumoperitoneum, ascites or an ingested button battery. o If contrast studies are obtained, water-soluble contrast agents are recommended because they are less irritating to the tissues in cases of perforation.

• Endoscopy: – Endoscopy helps to define the extent of the injury and develop a prognosis. Because of the unreliability of clinical findings in predicting significant esophageal injury, the threshold for endoscopy is low. – The optimal time for the procedure may be during the first several hours after the ingestion. It should be avoided during the subacute phase (5 to 15 days after ingestion), when the tensile strength of tissues is lowest.

– Any child with a history of significant ingestion, with oral lesions, or who is otherwise symptomatic, warrants endoscopy. – Evidence of perforation or shock is a contraindication to endoscopy. – Discontinue endoscopy immediately if esophageal burn is identified. – Repeat evaluation may be necessary after discharge if symptoms persist.

• Button or disk batteries: o These are highly corrosive and ingestion can lead to gastric burns, catastrophic erosion of esophagus and overlying major arteries. o If the initial CXR shows the battery in the esophagus, immediate endoscopic retrieval is indicated. o If the battery is beyond the esophagus, the patient may be discharged, with follow-up GI radiographic studies required if battery has not passed in 4 -5 days.

• Complications • Airway edema or obstruction may occur immediately or up to 48 hours following an alkaline exposure. • Gastroesophageal perforation acutely or later. – Deep circumferential or deep focal burns may result in strictures in more than 70% of patients; these strictures typically develop 2 -4 weeks post-ingestion. – Gastric outlet obstruction may develop 3 -4 weeks after an acid exposure. – Delayed upper GI bleeding may occur in acid burns 3 -4 days after exposure as the eschar sloughs. • Long-term risks include squamous cell carcinoma, which occurs in 14% of all significant exposures and may occur as late as 40 years after exposure.

• METHEMOGLOBINAEMIA
 is hemoglobin with the iron oxidized to the ferric")
• Methemoglobin (meth. Hb) is hemoglobin with the iron oxidized to the ferric (Fe 3+) state from the normal ferrous (Fe 2+) state. • Meth Hb is normally present in RBC at concentrations of 1 -2 % • Acquired methemoglobinemia can be induced by many drugs/chemicals, e. g. , chloroquine, dapsone, local anesthetics, high doses of methylene blue, metoclopramide, naphthalene, nitrates, nitrites, rifampin, toluidine, contaminated well water. • Infants are more sensitive than adults to methemoglobin-producing agents. • Consequences of methemoglobinemia: • Decreased oxygen binding and t impaired O 2 transport to tissues. • Shift of the O 2 dissociation curve to the left, thus increasing affinity of normal hemoglobin for oxygen, further compromising oxygen delivery to the tissues. • Agents causing methemoglobinemia can also cause hemolysis.

Clinical presentation: • Symptoms depends on level of amount of Meth. Hb • Clinical cyanosis may be seen with 1. 5 g/ d. L of meth. Hb (10% to 20% of total Hb), whereas it would take 5 g per d. L of deoxy-Hb to produce a similar degree of cyanosis. Patients with significant methemoglobinemia have a characteristic slate- gray cyanosis unresponsive to ↑ Fi. O 2. • Exposure of a drop of blood from patients who are cyanosed from cardiopulmonary disease will turn rapidly red after exposure to air (e. g. , by placing a drop of blood on a filter paper). However, blood with high levels of meth. Hb will remain dark on exposure to air • Pulse oximetry may be misleading (fixed at ≈ 85%) in patients with methemoglobinemia • Pulse oximetry may be misleading, pulse oximetry over-estimates the Sa. O 2 when < 85% and under-estimates the Sa. O 2 when > 85% so that the displayed Sp. O 2 is always in the range of 85% in methemoglobinemia despite much lower/greater levels of Sa. O 2.

Laboratory diagnosis: • Spectrophotometry will establish the diagnosis. A characteristic spectrometric peak of methemoglobin appears at 630 nm, which disappears after the addition of cyanide. • If the peak fails to disappear despite the addition of cyanide, sulfhemoglobinemia or hemoglobin M disorder should be suspected. • Quantitative methemoglobin can be measured with a cooximeter.

Management: • • • As in any case, ABCs takes the priority. Administer 100% oxygen; Supplemental oxygen can maximize tissue oxygen delivery. Note that pulse oximetry is not accurate. Gastrointestinal decontamination, in form of activated charcoal or gastric lavage may prevent further absorption of ingested methemoglobin-inducing agents. Asymptomatic or only mildly symptomatic patients with methemoglobin levels less than 30% should be observed for 24 to 48 hrs as this is the half life of methemoglobin, and just removal of offending agent may be adequate. If methemoglobin levels exceed 30 % or the patient exhibits clinical signs of hypoxia, administration of methylene blue is recommended. Dose: 1 -2 mg/kg of methylene blue may be diluted in 0. 9 % Na. Cl to concentration of 1 mg/ml and administered slowly IV over 5– 10 minute. Repeat after 1 hour if still symptomatic, severe cyanosis or methemoglobin >30%.

• Maximum dose: 7 mg/kg. If the offending drug has long half-life (e. g. , dapsone), give 0. 1 mg/kg/hr infusion. Contraindicated in G 6 PD deficiency. • Methylene blue can cause emesis, retrosternal chest pain, tachycardia, hypertension, anxiety, green-blue urine, oxidative hemolysis, headache, dizziness, factitious cyanosis. • In patients with G 6 PD deficiency or at higher doses, methylene blue can paradoxically induce methemoglobinaemia. • All measures to reduce oxygen demand should be employed; patients activity to be restricted to prevent symptom exacerbation. • Indications for exchange transfusions or hyperbaric oxygenation: Methemoglobin levels > 50 %, severe symptoms, those unresponsive to methylene blue, G 6 PD deficiency.

• Ascorbic acid is useful for congenital methemoglobinemia, it may have limited use, if any, in the treatment of acquired methemoglobinemia. • If exposure to a local anesthetic is the causative agent, an antidysrhythmic other than lidocaine should be used in case of arrhythmia. • Patient to be observed for 2 -3 days for hemolysis and should be advised to avoid further contact with the offending agent.

Methemo Clinical features globin % < 30 % No symptoms / mild acrocyanosis Treatment Observation, no treatment required 30 - 55 Poor feeding, lethargy, and % irritability. The older child may complain of fatigue, dizziness, headaches, and weakness. IV Methylene blue 1 -2 mg/kg every 5570% Methylene blue as above, exchange transfusion, hyperbaric oxygen > 70 % Potentially lethal. Respiratory depression, cardiac arrhythmias, seizures, and coma. 30– 60 min for severe cyanosis ( max 7 mg/kg) or till methemoglobin >30% or clinically better. Methylene blue, exchange transfusion, hyperbaric oxygen

• Organophosphate poisoning
 and. e les entre common ci chicken incriminated")
• orgaphosphates (OP, more potent) and. e les entre common ci chicken incriminated in poisoning episodes. Op poisonings in children occur via accidental dermal cantamination/ingestion. Suicidal are next more common • Signs and symptoms of toxicity appear when 50%-% ACHE activity has been inhibited • Enzyme regeneration occurs by either fresh synthesis of the enzyme or hydrolysis of the bond. • Therapy with oximes regenerate ACHE at the muscarinic and nicotinic synapses by reversing the phosphorylation of the enzyme before molecules become resistant to reactivation • Ingestions ate common in the adolescent. over 24 to 48 hours, mots ACHE beyond 12 to 24 hours after exposure to
 by phosphorylation of")
PHARMACOLOGY • OP compounds irreversible inhibition of the enzyme acetylcholinesterase (ACE) by phosphorylation of the active site. • Carbamates are transient inhibitors of ACHE, which bind to but they do not modify the enzyme, ACHE activity tered when the OP or carbamate spontaneously leaves the enzyme active site • Inhibition of ACHE allows acetylcholine to remain active in the synapse Resulting in sustained depolarization of the post synaptic neuron. • Effects occur via the nicotinic (CNS, autonomic ganglia, neuromuscular junction) of the Muscarinic receptors (secretomotor glands. smooth muscles) • Nicotinic sites are initially stimulated and then depressed hyperpolarization block). • Efects at muscarinic sites are sustained by continuous Simulation. • Sign and symptom appear when 50 -60 % of enzyme is inhibited

• Enzyme regeneration may occur by regeneration or hydrolysis. • Oxime regerarate by reversing the phosphorylation • Aging – after 24 -48 hours, ezmyme become resistant to reactivation by oxime. • Toxicity begins in minutes to hours. Delayed eg=ffect may be seen in fenthion or malathion. • Respiratory failure is most coomon cause of death – bronchospasm, ARDS, aspiration, CNS depression • Fasciculations een in severe poisoning.

Muscarinic effects : Pupils – miosis Ciliary body – blured vision Exocrine glands – bronchorrhea, rhinorrhea, salivation, tearing • Heart – bradycardia, dysrhyttmia • Smooth muscle bronchospasm , cramps, diarrhea, incontinence, panceartitis • • • Nicotinic effects : • Skeletal musclefasciculation followed by paralysis • Sympathetic ganglia – tachycardia , hypertension then hypotension • CNS – anxiety, restlessness, insomnia, tremor, ataxia, confusion, seizures and coma •

DIAGNOSIS • Diagnosis is based on a history of exposure, clinical findings, and improvement after appro priate antidotal therapy. Blood cholinesterase says may not be readily available. • CBC, serum electrolytes , creatinine low Cose, calcium, magnesium, lipasa, ABG and chest radiography for the initial work-up. (pseudocholinesterase) and red blood cell (RBC) acetylcholinesterase may be used to con firm the clinical diagnosis, if the tests are available if the clinical course is atypical • Plasma cholinesterase, butyrylcholinesterase and RBC cholinesterase level help in diagnosis

MANAGEMENT • Patients with severe toxicity should be admit ted to an ICU, • All personnel who are actively involved in the decontamination process should wear masks, • Clothing have to removed and body washed with soap and water • GI decontamination and activated charcoal is indicated. • Sorbitol may be used as cathartic.

• ABC stabilization • Indications for intubation Severe respiratory/ distress failure, laryngospasm, excessive bronchorrhea, low GCS and seizures • If intubation is necessary, succinylcholine should be avoided as it is metabolic by palsma Ache • IV boluses is needed • Pressors may be used • Benzodiazepines may be used for agitation or seizures

Antidotal therapy • Atropine is a competitive antagonist are acetylcholine at the muscarinic receptors but has no effect on nicotinic muscle weakness or paralysis. • It does not affect the ACh. E generation rate. • Atropine is indicated for control of pulmonary secretions and bronchospasm. It has a secondary role in helping control seizures and CNS manifestations of poisoning • Atropine therapy should be started at the first signs of cholinergic excess In general, more atropine is required during the first 24 hours. Requirement decreases when ACh is reactivated by 2 -PAM • The dose is 0. 05 mg kg in frequent s (every 10 min) as needed

• • The main end point of atropinization are resolution of bronchorrhea remember that focal crepitations or wheeze may be noted when there has been pulmonary piston) and normalization of heart rate, if bradycardia wan present. Other signs are pupils no longer pinpoint, the axilla is dry nomal systolic blood pressure. Mydriasis poor marker for adequate atropine. Presence of all signs is not mandatory to label patient as adequately atropinised If atropinization is not achieved by 3 -5 minutes, dose is doubled. Continue to double the dose and give IV every J-5 minutes until atropinization is achieved. Large doses hundreds of mg) may be required in some patients After atropinisation, maintain the effort by infusion (10%-20% of the total loading dose required for atropinisation) or with less frequent bolus doses A common pitfall is adequate atropine do ing. High doses of atropine are commonly needed for control of secretions. Regular observations are many to ensure maintenance of adequate atropinisation and prevent over atropinisation ( delirium, confusion, absent bowel sounds and urinary retentition) Incise of overatropinisation, stop the pine infusion. Check for resolution of si every 30 min. When they uttle, 80% of the previous rate.

• The patient should then be seen frequently to ensure that the new infusion rate has reduced the signs of atropine toxicity without resulting in the reappearance of cholinergic sign • Tachycardia is not a contraindication t - ing atropine. It may reflect hypoxia, hypovolemia, sympathetic stimulation etc • Glycopyrrolate (5 mg/kg) can be for atropine when isolated peripheral toxicity is present.
 • - PAM nucleophilic oxime that regenerates ACHE at the")
• Pralidoxime (PAM) • - PAM nucleophilic oxime that regenerates ACHE at the muscatine and nicotine sites by reversing phosphorylation de active site on the enzyme. • - PAM therapy is most effective when it early. Indications for PAM include • - Need to treat with atropine – • Muscle fasciculations • • Respiratory muscle or limb weakness • CNS effects.

• The ideal dosing of PAM is controversy – • Recent randomized control studies reviews suggest that higher dose ocim more efficacious than low dose, although on is discrepancy concerning the need for o all Whereas Indian studies now no advantage ing times pech Vietnamese model suggests that doses may be prevented. • Low dose PAM 20 -40 ngkg (max 1 gm) in N 130 ml NS over 30 minutes. This dosc an be repeated in I born if fasciculations or le weakness is still present. Subsequent doses: 20 -40 mg/kg Q 6 -8 hours. • WHO recommends 8 mg/kg/hour infusion or atematively, 30 mg/kg every 4 hours if continous infusion is not possible

• Empiric adjustments to this dosing regimen aybe necessary based on the patient's clinical response • duration of therapy is based on clinical game and is usually 24 to 48 hours Oximes be continued until recovery (12 hours her stopping administration of atropine or t rum cholinesterase level increases) therapy should be restarted if signs and symptoms recur • adverse effects – emesis, tachycardia laryngeal sapsm, muscle rigidity, transient neuromuscular blockade and respiratory/cardiac arrest, paradoxical ACh. E inhibition. • PAM may be less effective in late presenation • PAM may not be required in carbamate poisoning.

• Late EFFECTS OF OP POISONING Intermediate syndrome – 5 -10 percent High potency OP Recurrence or new onset weakness of neck muscles, proximal limb, respiratory muscle but without prominebt muscarinic signs Begin 24 -96 hours lasting 4 -18 days • • OPIDN - OP induced delayed peripheral neuropathy 1 -3 weeks after Sensory symptoms and later motoe involvement Recovery in 6 -18 months • • • Neuropsychiatric Anxiety, emotional lability, depression. Reolve by 1 year. Prognosis Severe OP poisoning may require prolonged respiratory support Carbamate has good prognosis. • • •

• Hydrocarbon Poisoning

• • Hydrocarbons are a group of organic compounds composed primarily of hydrogen and carbon. 3 basic types – Aliphatic (greatest risk for aspiration and pulmonary toxicity): Kerosene, lighter fluid, lubricating oils, gasoline. – Aromatic (systemic toxicity): Camphor, turpentine, benzene, toluene – Halogenated: Carbon tetrachloride, methylene chloride, trichloroethane. • • • Kerosene remains the commonest household hydrocarbon involved in accidental ingestion. Others include paint thinners/ removers, petrol, diesel, turpentines. Most exposures occur in younger children and involve accidental ingestions. Storage in unmarked, readily accessible containers accounts for the high exposures in young children. In adolescents and adults, poisoning generally results from inhalational abuse, occupational exposure, intentional ingestion, or accidental aspiration during the siphoning of fuels.

• • • • Pathophysiology: Systemic toxicity is limited by poor GI absorption. After ingestion, the major toxicity is the potential to cause a fulminant and sometimes fatal chemical pneumonitis. CNS, GI, CVS, hepatic, renal, hematologic, and cutaneous toxicities may also occur. Pulmonary effects: Pulmonary toxicity results directly from aspiration rather than from hematogenous spread. Aspiration can occur even without vomiting. Even small quantities (<1 m. L) of low viscosity, low surface tension and high volatility aliphatic hydrocarbons can result in significant injury. Occasionally, less inflammatory but more localized and indolent lipoid pneumonia may occur. CNS effects: The most common CNS symptoms include headache, lethargy, seizures and coma. Nonspecific symptoms such as weakness and fatigue may also be reported. Cardiac effects: Major dysrhythmias may occur. Etiologies include hypoxia, myocardial sensitization to catecholamines and direct myocardial damage. Other effects: Bone marrow toxicity and hemolysis. Hepatic and renal failure, including renal tubular acidosis may occur with chlorinated hydrocarbons. Direct contact with the skin and mucous membranes may cause local irritation or even extensive chemical burns. GI irritation following ingestion results in nausea, vomiting, sore throat. Vomiting increases the likelihood of pulmonary aspiration.

• • • • Clinical Manifestations: Patients who aspirate generally demonstrate symptoms within 30 minutes. Those who do not have symptoms after 6 hours of exposure remain asymptomatic. Initial coughing, gasping, and choking may progress and peak during the first 24 to 48 hours with tachypnea with grunting respirations, nasal flaring, retractions, and cyanosis: some may develop pulmonary edema and hemoptysis. The odor of kerosene or petroleum may be apparent. Within the first 24 to 48 hours, fever and leukocytosis are common. The persistence of fever beyond 48 hours may suggest a bacterial super-infection. In most cases, symptoms resolve during the next 2 to 5 days with supportive care. The severity of CNS dysfunction often correlates with the severity of aspiration. Cardiovascular toxicity is uncommon, but dysrhythmias and sudden death after gasoline siphoning have been reported. Radiographic abnormalities occur in up to 75% of hospitalized patients, appearing within 2 hours in 88% of patients and by 12 hours in 98%. Common findings include fine perihilar opacities, bilateral basilar infiltrates, and atelectasis. CXR findings correlate poorly with clinical symptoms, lag behind clinical improvement, and may persist for several days to weeks after symptoms have resolved. Initial blood gases may demonstrate hypoxemia and hypocarbia: this can progress to hypercarbia and acidosis. Acute toluene intoxication results in a wide anion gap acidosis. The presence of a profound wide anion gap acidosis, especially in a patient appearing intoxicated, should prompt an evaluation for other etiologies (eg, methanol, ethylene glycol, salicylates).

• • • Management: Asymptomatic patients should be observed with continuous pulse-oximetry for a period of at least 6 hours. If the patient remains asymptomatic (eg, no coughing, vomiting, tachypnea, or other evidence of respiratory difficulties), then a chest radiograph may be obtained to evaluate for aspiration. Patients with ingestions who remain or become asymptomatic with a normal chest radiograph (obtained 6 hours or more after exposure) may be discharged after 6 hours of observation. All symptomatic patients, those with abnormal chest radiographs, arterial blood gases, or pulse oximetry and patients with suicidal intent should be hospitalized. Airway, breathing, and circulation: Stabilization of the airway is always the first priority in any intoxication. Give supplemental oxygen to all patients and perform beside pulse oximetry. Early intubation, mechanical ventilation, and use of positive end-expiratory pressure may be indicated in patients with severe hypoxemia, respiratory distress or a decreased conscious level. Decontamination

• Avoid emesis or lavage because of the risk of aspiration and minimal risk of systemic absorption. • Consider lavage only after intubation with cuffed ETT if the hydrocarbon contains a potentially toxic substance (e. g. , insecticide, heavy metal, camphor) and a toxic amount has been ingested. • Avoid activated charcoal unless there is co-ingestion. It does not bind aliphatics and will increase the risk of aspiration. • Cutaneous decontamination in cases of cutaneous exposure: Remove contaminated clothing and thoroughly wash the skin with soap and water. Vapor inhalation and cutaneous absorption may occur long after the exposure. • Arrhythmia: Correct abnormal electrolyte levels (especially magnesium and potassium). Avoid catecholamines, lidocaine or beta-blockers may be useful. Involve cardiology.

• • • Supportive care in the PICU: Antacids and acid suppression for GI symptoms. Beta-agonist nebulizations for bronchospasm. Baseline renal and liver function studies and a toxic screen should be obtained if toxic additives or concomitant ingestion is suspected. In patients with pneumonitis, monitoring includes serial evaluation of acid-base, electrolyte and fluid balance (e. g. , patient to be kept “dry” after perfusion normalizes) and CXR. No role for steroids. Antibiotics should be given only to patients with documented bacterial pneumonias (e. g. , Gram's stain or culture of sputum or tracheal aspirate), worsening CXR, leukocytosis or fever persisting beyond the first 48 hours. Positive airway pressure may be necessary to maintain oxygenation, monitor for air-leaks. Rescue therapies for refractory hypoxemia include high-frequency ventilation, surfactant and extracorporeal membrane oxygenation. Most patients with petroleum distillate poisoning recover fully with supportive care

• TCA antidepressant intoxication

Clinical features Neurologic Sedation, coma, seizures Cardiac Tachycardia, hypotension, conduction abnormalities Anticholinergic Dilated pupils, dry mouth, absent bowel sounds, urinary retention

Diagnostic evaluation Electrocardiographic changes in severe poisoning: QRS duration >100 msec Rightward deflection of the terminal 40 msec of the QRS complex Deep S wave in leads I, AVL; tall R wave in lead AVR R wave in AVR >3 mm; R/S ratio in AVR >0. 7 Serum TCA concentrations do not help to guide therapy

Treatment Airway Manage as indicated; many patients require tracheal intubation Breathing Administer supplemental oxygen Circulation Hypotension: Treat with intravenous boluses of isotonic crystalloid. If patient remains hypotensive despite aggressive volume resuscitation, can treat with a vasopressor. Alpha-adrenergic agonists (eg, neosynephrine, norepinephrine) are preferred. Conduction disturbances: If QRS >100 msec, challenge with intravenous sodium bicarbonate (2 to 3 m. Eq/kg up to 150 m. Eq IV push) and assess for QRS narrowing. If QRS narrows, begin continuous infusion (150 m. Eq of sodium bicarbonate in 1 liter of D 5 W to run at 250 m. L/hour in adults or twice the maintenance fluid rate in children).

Gastrointestinal decontamination Administer activated charcoal if patient presents within 2 hours of ingestion, unless gastrointestinal complication (ileus, obstruction) suspected. Dose is 1 g/kg (maximum dose 50 g). Seizures Treat with benzodiazepines (eg, diazepam 5 mg IV or lorazepam 2 mg IV) Do NOT treat with phenytoin

• Anticholinergic poisoning

Clinical and laboratory features Anticholinergic toxicity is almost always a clinical diagnosis Manifestations of anticholinergic toxicity include: Flushing due to cutaneous vasodilation ("red as a beet") Anhydrosis ("dry as a bone") Hyperthermia due to loss of sweating ("hot as a hare") Blurry vision due to nonreactive mydriasis and paralysis of accommodation ("blind as a bat") Agitated delirium ("mad as a hatter") Urinary retention ("full as a flask") Decreased bowel sounds Tachycardia

Diagnosis Check fingerstick glucose, ECG, acetaminophen and salicylate levels, and a qualitative pregnancy test in poisoned patients A serum creatine kinase and renal function testing (BUN and creatinine) are appropriate in patients in whom rhabdomyolysis is suspected No laboratory findings or diagnostic laboratory tests can definitively determine anticholinergic toxicity A trial of physostigmine (see Treatment below) will help establish or rule out the diagnosis of anticholinergic toxicity

Treatment Secure the airway, breathing, and circulation Patients who manifest both peripheral AND moderate central (moderate to severe agitation/delirium) anticholinergic toxicity, without contraindications to physostigmine, should be treated with this medication; dose: 0. 5 to 2 mg (0. 02 mg/kg IV, up to a maximum of 0. 5 mg per dose in pediatric patients); physostigmine should be given by slow IV push, generally over five minutes Treat agitation and seizures with benzodiazepines (eg, lorazepam 1 to 2 mg IV push [pediatric dose 0. 1 mg/kg up to 2 mg maximum single dose]; may repeat as needed); DO NOT use phenothiazines or butyrophenones (eg, haloperidol) Give activated charcoal (1 g/kg; maximum 50 g) to patients with intact mental status or a secure airway and likely ingestion of an anticholinergic agent

• Salicylate poisoning

Clinical and laboratory features Common: tachypnea, tinnitus, nausea, vomiting, acidbase abnormalities Severe cases: hyperthermia, altered mental status, pulmonary edema
, basic electrolytes, BUN and creatinine,")
Diagnostic evaluation Plasma salicylate concentration, arterial blood gas (ABG), basic electrolytes, BUN and creatinine, chest radiograph Repeat salicylate concentration every two hours until it is declining Repeat ABG every two hours until acid-base status stable or improving

Treatment Avoid intubation if at all possible Administer supplemental oxygen as needed Volume resuscitate unless cerebral or pulmonary edema is present Administer multiple doses of activated charcoal (first dose: 1 g/kg orally up to 50 g) Administer supplemental glucose in patients with altered mental status, even if serum glucose concentration is normal: IV dextrose 50 g as 100 m. L of 50 percent dextrose

Alkalinize with sodium bicarbonate Bolus therapy: sodium bicarbonate, 1 to 2 m. Eq/kg (maximum 100 m. Eq) IV push over 3 to 5 minutes Maintenance therapy: 100 to 150 m. Eq sodium bicarbonate in 1 L of D 5 W, run at 250 m. L/hour in adults OR run at 1. 5 to 2 times maintenance in children Correct hypokalemia, hypocalcemia and other electrolyte abnormalities. IV sodium bicarbonate is NOT compatible with calcium salts. Alkalemia (arterial p. H up to 7. 55) is NOT a contraindication to sodium bicarbonate therapy DO NOT USE ACETAZOLAMIDE TO ALKALINIZE THE URINE

Alert nephrology team early in the patient's clinical course; indications for hemodialysis include: Profoundly altered mental status Pulmonary or cerebral edema Renal insufficiency that interferes with salicylate excretion Fluid overload that prevents the administration of sodium bicarbonate A plasma salicylate concentration >100 mg/d. L (7. 2 mmol/L) in acute ingestion OR >60 mg/d. L (4. 3 mmol/L) in chronic ingestion Clinical deterioration despite aggressive and appropriate supportive care

• Pesticide poisoning

• Organophosphate compounds inhibit acetylcholinesterase resulting in acute toxicity. Intermediate syndrome can develop in a number of patients and may lead to respiratory paralysis and death. Management consists of proper oxygenation, atropine in escalating doses and pralidoxime in high doses.

• Organochlorine pesticides are toxic to the central nervous system and sensitize the myocardium to catecholamines. Treatment involves supportive care and avoiding exogenous sympathomimetic agents.

• paraquat causes severe inflammation of the throat, corrosive injury to the gastrointestinal tract, renal tubular necrosis, hepatic necrosis and pulmonary fibrosis. Administration of oxygen should be avoided as it produces more fibrosis. Use of immunosuppressive agents have improved outcome in patients with paraquat poisoning.
.")
• Rodenticides include thallium, superwarfarins, barium carbonate and phosphides (aluminium and zinc phosphide). Alopecia is an atypical feature of thallium toxicity. Most exposures to superwarfarins are harmless but prolonged bleeding may occur. Barium carbonate Ingestion cause severe hypokalaemia and respiratory muscle paralysis.

• Aluminium phosphide is a highly toxic agent with mortality ranging from 37% to 100%. It inhibits mitochondrial cytochrome c oxidase and leads to pulmonary and cardiac toxicity. Treatment is supportive with some studies suggesting a beneficial effect of magnesium sulphate.

• Aluminium phosphide is a highly toxic agent with mortality ranging from 37% to 100%. It inhibits mitochondrial cytochrome c oxidase and leads to pulmonary and cardiac toxicity. Treatment is supportive with some studies suggesting a beneficial effect of magnesium sulphate.

• Ethylene dibromide-a highly toxic, fumigant pesticide-produces oral ulcerations, followed by liver and renal toxicity, and is almost uniformly fatal.
- Slides: 90