Chapter 5 Nucleophilic Substitution Advanced Organic Chemistry Chapter
 sh. Javanshir")
Chapter 5 Nucleophilic Substitution Advanced Organic Chemistry (Chapter 5) sh. Javanshir

 Mechanism First order, SN")
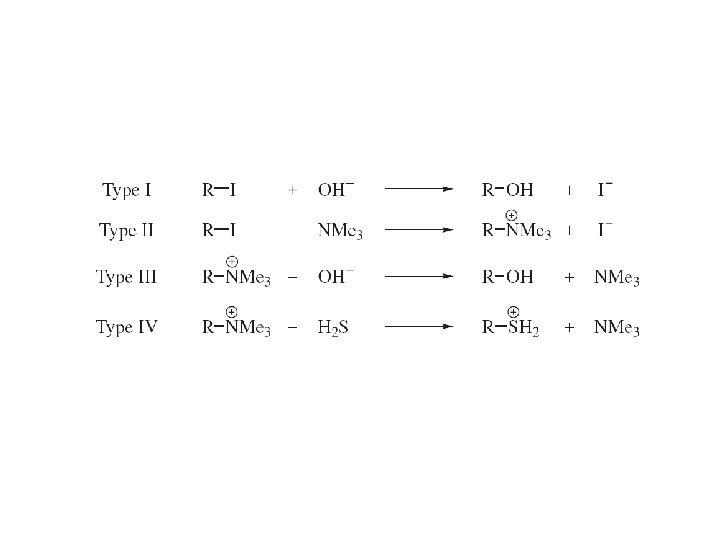
5. 1 The Limiting Case-Substitution by the Ionization (SN 1) Mechanism First order, SN 1 leaving group initiates reaction rate DOES NOT depend on nucleophile concentration

Fig. 5. 1. Reaction energy profile for nucleophilic substitution by the ionization SN 1 mechanism. ﺣﺎﻟﺖ گﺬﺍﺭ ﻃﺒﻖ پﺬیﺮﻩ ﻫﺎﻣﺖ ﺷﺒیﻪ ﺑﻪ ﺣﺪ ﻭﺍﺳﻂ کﺮﺑﻮکﺎﺗیﻮﻧی ﺍﺳﺖ
 Mechanism ﺑﺮ ﻫﻤکﻨﺶ ﻗﻮی")
5. 2 The Limiting Case-Substitution by the Direct (SN 2) Mechanism ﺑﺮ ﻫﻤکﻨﺶ ﻗﻮی ﺑیﻦ ﺍﻭﺭﺑیﺘﺎﻝ ﻫﺎ
 in the back-side attack. Side attack")
HOMO/LUMO interaction (n/σ*) in the back-side attack. Side attack

ﻭﺍﺭﻭﻧگی پیکﺮﺑﻨﺪی Fig. 5. 3. Reaction energy profile for nucleophilic substitution by the direct displacement SN 2 mechanism. The HOMO at the T. S. is p in character, therefore T. S. energy should be lowered by conjugation with adjacent substituents.

MO Description

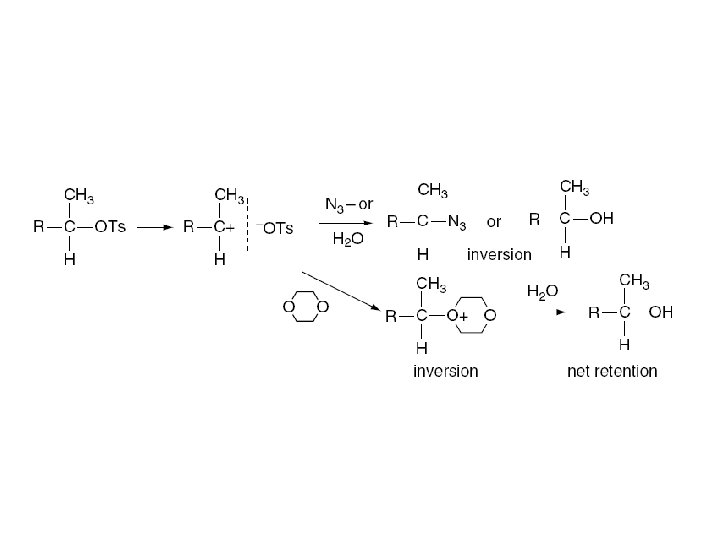
• Although many first-order substitutions do give complete racemization, many others do not. Typically there is 5– 20% inversion, although in a few cases, a small amount of retention of configuration has been found. These and other results have led to the conclusion that in many SN 1 reactions at least some of the products are not formed from free carbocations but rather from ion pairs.

5. 3 Detailed Mechanistic Description and Borderline Mechanisms Winstein: Concept of ion pairs ﻣﻔﻬﻮﻡ ﺟﻔﺖ یﻮﻥ ﺩﺭ ﻣکﺎﻧیﺴﻢ . ﻣﺮﺯی ﺣﺎیﺰ ﺍﻫﻤیﺖ ﺍﺳﺖ contact or tight ion pair Attacking the Nuclephile or Solvent: Intimate Ion Pair Inversion of Configuration Solvent Separated Ion Pairs Dissociated Ions Partial Racemization

The ion-pair concept thus predicts that SN 1 reactions can display either complete racemization or partial inversion.

Intimated and Solvent Ion Pairs: 80% acetone and 20% water ﺗﺒﺎﺩﻝ ﻋﻨﺼﺮ ﻧﺸﺎﻥ ﺩﺍﺭ p-chlorobenzhydril p-nitrobenzoate Dissociated Ions: The fact that the rate of isotopic exchange exceeds that of racemization indicates that ion pair collapse occurs with predominant retention of configuration. ﺩﺭﺟﻪ 100 ﺩﺭ
: kex: Unchanged")
Addition of nuclephile to the system (0. 14 M Na. N 3): kex: Unchanged krac: No Racemization The intermediate that can racemize is captured by azide ion and converted to substitution product with inversion of configuration. Slide 8

Isotope Labeling reveals Bond breaking without net substitution about one-fifth of the ion pairs recombine rather than react with the nucleophile. A similar experiment in acetic acid indicated about 75% internal return. Ion pair formation and recombination is occurring competitively with ion pair formation and substitution.

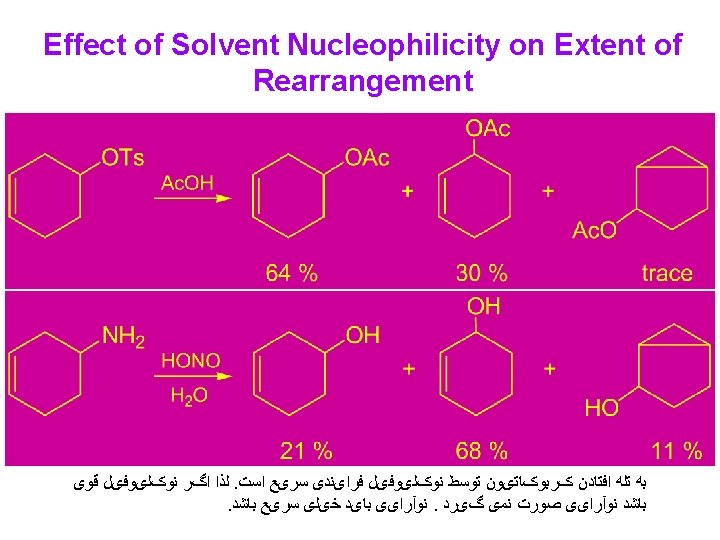
A study of the exchange reaction of benzyl tosylates during solvolysis in several solvents showed that with electron-releasing group (ERG) substituents, e. g. , p-methylbenzyl tosylate, the degree of exchange is quite high, implying reversible formation of a primary benzyl carbocation. For an electron-withdrawing group (EWG), such as m- Cl, the amount of exchange was negligible, indicating that reaction occurred only by displacement involving the solvent. When an EWG is present, the carbocation is too unstable to be formed by ionization. This study also demonstrated that there was no exchange with added “external” tosylate anion, proving that isotopic exchange occurred only at the ion pair stage.
 is")
Demonstrating the ion pair return The rate of decrease of optical rotation (racemisation) is greater than the rate of product formation. The solvent-separated ion pair is the most likely intermediate to play this role.

. ﺭﺍﺳﻤیک ﺷﺪﻥ ﻫﻤﻮﺍﺭﻩ ﺑﺎ ﺗﺒﺎﺩﻝ ﺍیﺰﻭﺗﻮپی ﻫﻤﺮﺍﻩ ﻧیﺴﺖ sulfonate can rotate with respect to the carbocation without migrating to its other face. The unlikely alternative ﻣکﺎﻧیﺴﻢ ﻫﻤﺎﻫﻨگ

Special Salt Effect: • Addition of salts typically causes an increase in the rate of solvolysis of 2° alkyl arensulfonates that is linear with salt concentration (due to the increase in dielectric constant of the medium). • The addition of Li. Cl. O 4 or Li. Br in the acetolysis of certain tosylates produced an initial steep rate acceleration that then decreased to the normal linear acceleration (caused by the ordinary salt effect). • This is interpreted as follows: the Cl. O 4 - (or Br-) traps the solvent separated ion pair to give R+ Cl. O 4 - which, being unstable under these conditions, goes to product. Hence, the amount of solvent-separated ion pair that would have returned to the starting material is reduced, and the rate of the overall reaction is increased.

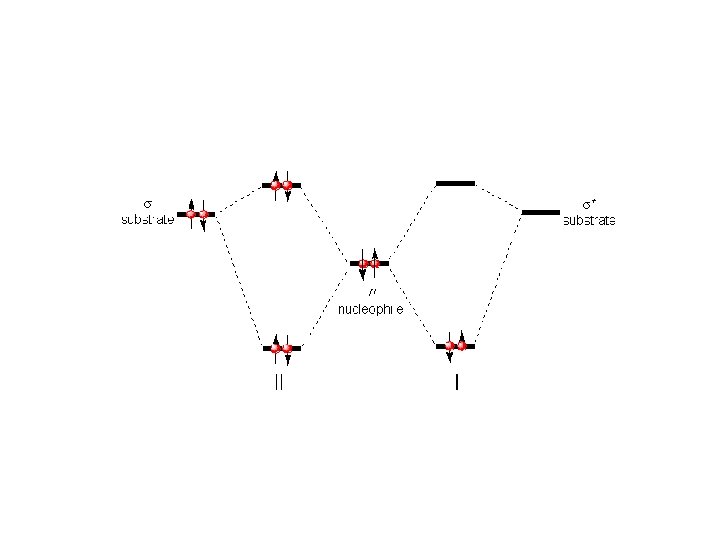
Robbins: Ion pairs might not only be involved in SN 1 and borderline processes but also in displacement exhibiting the stereochemical and kinetic characteristic of the SN 2 process. Nu Attack Solvent Attack R+ X - Inversion R+ ║ X - Inversion Retention or Inversion Racemization R+ + X -

Fig. 5. 4. Schematic relationship between reactants, ion pairs, and products in substitution proceeding through ion pairs.

Fig. 5. 6. Reaction energy profiles showing decreasing carbocation stability in change from SN 1(lim) to SN 2(lim) mechanisms.
 Solvent nucleophilicity")
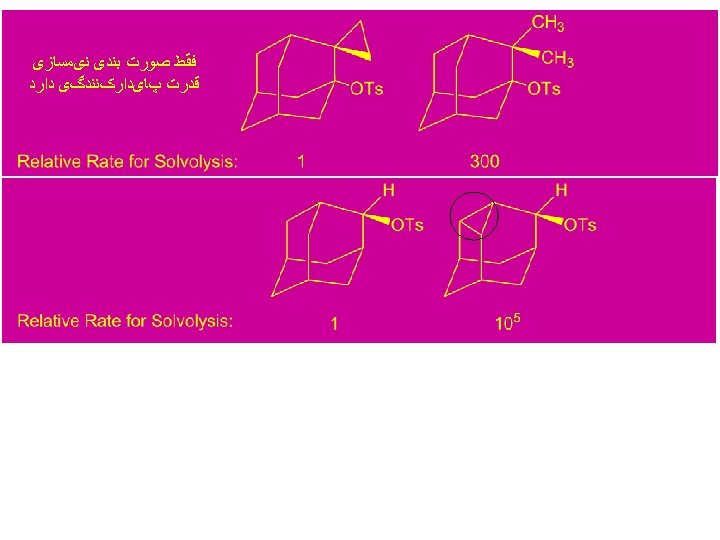
Nucleophilic participation of the solvents in borderline mechanisms is related to: a) Solvent nucleophilicity Solvents like CF 3 COOH or CF 3 CH 2 OH are used to define the characteristics of reactions proceeding without nucleophilic solvent participations b) Structure of the reactant (2 - adamantyl system has been considered as a model reactant for defining of ionization without nucleophilic solvent participations) Solvation is minimized by steric hindrance and the 2 -adamantyl system is regarded as being a secondary reactant that cannot accommodate significant back-side nucleophilic participation.
")
5. 4 Carbocations گﺮﻣﺎگیﺮ Ionization in solvent is feasible because of solvation. Evidences: 1) solution of Ph 3 CCl (trityl chloride) in liquid SO 2 is conducting. 2) Ph 3 CCl. O 4 has ionic behavior. Triphenyl methyl chloride

Relative Stability HR: Acidity function of the medium For dilute solutions: HR = p. H Advanced Organic Chemistry (Chapter 5) sh. javanshir

Table 5. 1. Values of p. K R+ for Some Carbocations

Hydride Affinity Carbocation stability in the gas phase can be measured by mass spectrometry and reported as hydride affinity, which is the enthalpy of the reaction: Table 5. 5. Hydride Affinity of Some Carbocations

Stability Order of Carbocations Based on Solvolysis Rate: 3° > 2° > 1° > CH 3+ Stabilization of Carbocations: ﺳﺪ ﺍﻧﺮژی چﺮﺧﺸی

The destabilizing effects of CYANO and FORMYL groups are less. They can act as a p donors.

Table 5. 1. Values of p. K R+ for Some Carbocations

Cyclopropyl Cation: tri-Cyclopropyl methyl Cation > tri-phenyl methyl cation ﺻﻮﺭﺕ ﺑﻨﺪی ﻧیﻤﺴﺎﺯی
: FSO 3")
Study of Carbocations Rearrangement NMR spectroscopy in super acid media (magic acid): FSO 3 H - Sb. F 5 - SO 2 Powerful protonating ability & isobutanol FSO 3 H - Sb. F 5 - SO 2 o -30 C Non-Nucleophilic t-Bu+ ( کﺘﺎﺏ 3 -5 )ﻃﺮﺡ. یﻮﻧﺶ ﺩﺭ ﺳﻮپﺮ ﺍﺳیﺪ ﻫﺎ ﻣﻨﺠﺮ ﺑﻪ ﺗﺸکیﻞ پﺎیﺪﺍﺭ ﺗﺮیﻦ کﺮﺑﻮکﺎﺗیﻮﻥ ﻫﺎ ﻣی ﺷﻮﺩ Rearrangement: → t-Bu C → t-pentyl C → t-hexyl C 4 + 5 6 + +
 Decrease of rate of solvolyse Increase of hydride")
Carbocations have Sp 2 hybridization (planar) Decrease of rate of solvolyse Increase of hydride affinity
 parameter thermodynamic (or")
NUCLEOPHILES AND BASES THE FUNDAMENTAL DISTINCTION Nucleophilicity Basicity kinetic (or rate) parameter thermodynamic (or equilibrium) parameter. All nucleophiles are bases …. . . and all bases are nucleophiles. HOWEVER : A good base is not necessarily a good nucleophile, and vice versa.
![NUCLEOPHILE VERSUS BASE Nu 1 Nucleophilicity = Kinetic Rate = k 2[RX][Nu] Nu 2](http://slidetodoc.com/presentation_image_h/91c922700c45a9aef17b6630d67737a5/image-37.jpg "NUCLEOPHILE VERSUS BASE Nu 1 Nucleophilicity = Kinetic Rate = k 2[RX][Nu] Nu 2")
NUCLEOPHILE VERSUS BASE Nu 1 Nucleophilicity = Kinetic Rate = k 2[RX][Nu] Nu 2 is a better nucleophile ( FASTER RATE ) Nu 2 good nucleophile increases k 2 (I. e. , the rate) Basicity = Thermodynamic B: - + H+ B-H strong base shifts equilib. Nu 1 is a better base ( STRONGER BOND )

DIFFERENT PLACES ON THE ENERGY PROFILE DETERMINE NUCLEOPHILICITY AND BASICITY NUCLEOPHILES Nucleophilicity is determined here activation energy and rate (kinetics) faster is better BASES Basicity is determined here strength of bonds and position of equilibrium lower energy is better
 A high")
5. 5 Nucleophilicity and Solvent Effect Factors the Effect on Nucleophilicity: 1) A high Solvation Energy of The Nuclephile lowers the G. S. and increase the activation energy. 2) Stronger bond between nucleophilic atom and carbon cause the stabilization of the T. S. and will reduce the activation energy. 3) A bulky nuclephile will be less reactive than smaller one because of non-bonded repulsions that develop in the T. S. 4) High electronegativity is unfavorable. 5) Polarizibility: The more easily distorted the atom, the better its nucleophilicity. Polarizibility increase with atomic number going down in the periodic table.

WHAT IS THE IDEAL NUCLEOPHILE ? LARGE . . : Y : . . SN 2 REACTIONS STERIC PROBLEMS no way ! bad R SMALL . . : . . X: C good : Br R Smaller is better ! R For an SN 2 reaction the nucleophile must find the back lobe of the sp 3 hybrid orbital that the leaving group is bonded to.

EXPECTED “IDEAL” NUCLEOPHILES cyanide ROD OR SPEAR - : C N: SHAPED + : N N N: azide These types should be able to find the target ! SMALL SPHERES . . : Cl: : F: . . Generally this idea is correct. etc.

OUR NAÏVE EXPECTATION We would expect the halides to be good nucleophiles: ionic radii: 1. 36 A 1. 81 A F- Cl - 1. 95 A 2. 16 A smallest ion Br - I- and we would expect the smallest one (fluoride) to be the best nucleophile, …. . however, that is not usually the case.

EXPERIMENTAL RESULTS RELATIVE RATES OF REACTION FOR THE HALIDES CH 3 -I + Na. X Me. OH CH 3 -X + Na. I Rate = k [CH 3 I] [X ] k F- 5 x 102 Cl- 2. 3 x 104 Br- 6 x 105 I- 2 x 107 slowest fastest * Me. OH solvates like water but dissolves everything better. S N 2

SOLVATION Solvation reverses our ideas of size.

HEAT OF SOLVATION ENERGY IS RELEASED WHEN AN ION IS PLACED IN WATER F- gas phase - 120 Kcal / mole HEAT OF SOLVATION F- (g) O H H H O F- H H O H SOLVATED ION water solution F- (aq) The interaction between the ion and the solvent is a type of weak bond. Energy is released when it occurs. Solvation lowers the potential energy of the nucleophile making it less reactive.

HALIDE IONS IONIC RADIUS smallest ion Heats of solvation in H 2 O 1. 36 A 1. 81 A 1. 95 A F Cl - Br - - 120 - 90 - 75 2. 16 A I- - 65 Kcal / mole - X(H 2 O)n larger n increasing solvation smaller n SMALL IONS SOLVATE MORE THAN LARGE IONS

SMALL IONS SOLVATE MORE HEAVILY THAN LARGE ONES F strong interaction with the solvent O H H H O O H H I BETTER NUCLEOPHILE H O - H O H solvent shell “Effective size” is larger. Heavy solvation lowers the potential energy of the nucleophile. It is difficult for the solvated nucleophile to escape the solvent shell. This ion is less reactive. - H O H H . . . smaller solvent shell. . . escapes easily …more potential energy weak interaction with the solvent

PROTIC SOLVENTS water methanol amines Water is an example of a “protic” solvent. Protic solvents are those that have O-H, N-H or S-H bonds. Protic solvents can form hydrogen bonds and can solvate both cations and anions.

LARGER IONS ARE BETTER NUCLEOPHILES IN PROTIC SOLVENTS THREE FACTORS ARE INVOLVED : 1 2 3 In protic solvents the larger ions are solvated less (smaller solvent shell) and they are, therefore, effectively smaller in size and have more potential energy. Since the solvent shell is smaller in a larger ion it can more easily “escape” from the surrounding solvent molecules during reaction. There is more potential energy. The larger ions are thought (by some) to be more “polarizable”. see the next slide …. .

POLARIZABILITY Polarizability assumes larger ions are able to easily distort the electons in their valence shell, and that smaller ions cannot. C Br VERY HYPOTHETICAL The distortion of large ions is easier because the orbital clouds are more diffuse. The nucleophile “flows” into the reactive site.

Polarizability Effect 51

BASICITY If everything else is equal, the stronger base is the better nucleophile. This principle shows up in a period, where atoms do not vary appreciably in size, and solvate to similar extents. OH- is a better nucleophile than F-
: Reference Reaction: Methanolysis of Me-I")
Nucleophilicity Constant (n): Reference Reaction: Methanolysis of Me-I

Table 5. 7. Nucleophilicity Constants for Various Nucleophiles

Nucleophilicity and Basicity Relationship The correlation is better if the attacking atom is the same CH 3 O- > Ph. O- > CH 3 COO- > NO 3 n= 6. 3 5. 8 4. 3 1. 5 Pka= 15. 7 9. 89 4. 8 -1. 3 Nucleophilicity increase going down the periodic table. I- > Br- > Cl- > FPh. Se- > Ph. S- > Ph. O-

Hard-Soft-Acid-Base Concept Hard nucleophiles prefer hard electrophiles while soft nucleophiles prefer soft electrophiles. The sp 3 carbon is a soft electrophile, whereas the proton is a hard electrophile Therefore, a soft anion should act as a nucleophile, nucleophile giving the substitution product, while a hard anion is more likely to abstract a proton, giving the elimination product

Table 5. 8. Hardness and Softness of Some Common Ions and Molecules

Scheme 5. 1. Examples of Competition between Nucleophilicity and Basicity

a- Effect Atoms which are directly bonded to an atom with one or more unshared pairs of electrons tend to be stronger nucleophiles than would otherwise be expected. HOO- > HO- H 2 NNH 2, NH 2 OH > NH 3 1) G. S. (ground state) Destabilization of the nucleophile by lone pair-lone pair repulsion 2) Stabilization of charge deficiency at the T. S. by lone pair.

Nucleophilicity order In protic solvents, e. g. Me. OH: - - I > Br > Cl - Anions are strongly solvated Greater nucleophilicity of soft anion Nucleophilicity order In polar aprotic solvents, e. g. , DMSO: - - I < Br < Cl - Anions are weakly solvated Greater nucleophilicity of soft anion Aprotic Solvents Cations are strongly solvated in aprotic solvents N-Methylpyrrolidone

APROTIC SOLVENTS - + dimethylsulfoxide “DMSO” + dimethylformamide “DMF” hexamethylphosphoramide “HMPA” acetone if scrupulously free of water acetonitrile APROTIC SOLVENTS DO NOT HAVE OH, NH, OR SH BONDS They do not form hydrogen bonds.
 + crowded - + The nucleophile")
APROTIC SOLVENTS SOLVATE CATIONS, BUT NOT ANIONS (NUCLEOPHILES) + crowded - + The nucleophile is “free” (unsolvated), and therefore is small and not hindered by a solvent shell.
")
DIMETHYLFORMAMIDE X nucleophile is “free” (unsolvated)

WHY NOT ALWAYS USE APROTIC SOLVENTS FOR SN 2 ? Mostly, it is a matter of expense. Water, ethanol, methanol and acetone are much cheaper, especially water. Water Methanol Ethanol Acetone “free” $14. 70 / L $15. 35 / L $16. 60 / L DMSO$47. 50 / L DMF $33. 75 / L HMPA $163. 40 / L Cheapest grades available, Aldrich Chemical Co. , 2000.


5. 6 Leaving Group Effects The reactivity of the leaving groups generally parallel their electron-attracting capacity. - CF 3 COO >> CH 3 COO - The order of reactivity of the halide leaving group (C-X bond): I > Br > Cl >> F C-F ﺑﺮﺍی kcal 100 ﺗﺎ C-I ﺑﺮﺍی kcal 50 ﺍﺯ : C-X ﻗﺪﺭﺕ پیﻮﻧﺪ Increasing the reactivity by coordination to electrophilic species: kcal/mol 37 =( ﺍﻧﺮژی ﻓﻌﺎﻟﺴﺎﺯی) ﺩﺭ ﻏیﺎﺏ ﺍﺳیﺪ The reaction is greatly accelerated in acidic media.

The Leaving Group u The more stable the anion, the better the leaving ability • the most stable anions are the conjugate bases of strong acids

Table 5. 12. Relative Solvolysis Rates of 1 -Phenylethyl Esters and Halides

5. 7 Steric and Strain Effects on Substitution In primary alkyl substrates the reaction rate decrease with increasing substrate size (direct displacement). Steric repulsions at T. S Table 4. 9. Rate Constants for Nucleophilic Substitution of Primary Alkyl Bromides and Tosylates ﺩﺭ ﻣﻮﺭﺩ ﺣﻼﻝ کﺎﻓﺖ ﺗﻮﺳیﻼﺕ ﺩﺭ ﺍﺳیﺪ ﺍﺳﺘیک ) ﻣﻮﺭﺩ چﻬﺎﺭﻡ (ﺣﺴﺎﺳیﺖ ﺑﻪ ﺍﺛﺮﺍﺕ ﻓﻀﺎیی کﻤﺘﺮ ﺍﺳﺖ چﻮﻥ . ﻧﻮکﻠیﻮﻓیﻞ ﻣﺸﺎﺭکﺖ کﻤﺘﺮی ﺩﺍﺭﺩ ﻭ پیﻮﻧﺪ آﻦ ﺑﺎ کﺮﺑﻦ ﺩﺭ ﺣﺎﻟﺖ گﺬﺍﺭ ﺳﺴﺖ ﺗﺮ ﺍﺳﺖ

In the case of ionization and stabilization of cationic T. S. , the reaction rate increase with increasing steric factor. Acetolysis: A high CH 3/H rate ratio is expected if nucleophilic participation is weak and stabilization of the cationic nature of the TS is important.
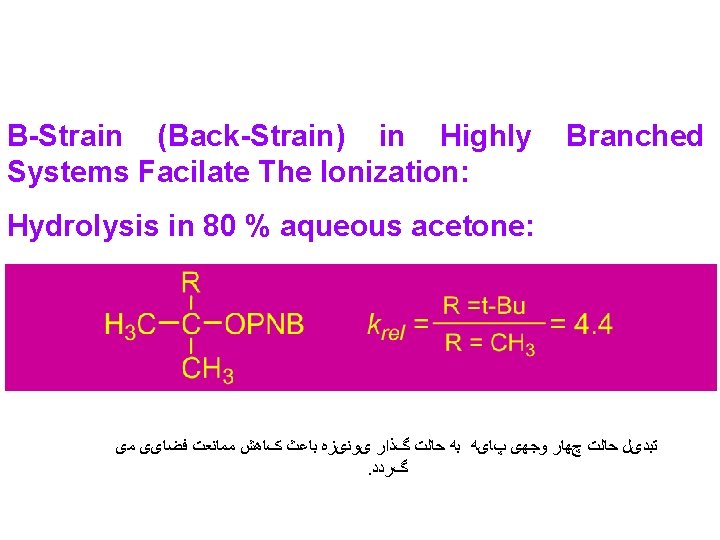

Large Back strain

5. 8 Substituent Effect on Reactivity a-Substituent effect: Direct SN 2 reaction proceeds more easier than SN 1 reaction in a-halo derivatives of ketones, aldehydes, acids, esters, nitriles and related compounds. Table 5. 15. Substituent Effects of -EWG Substituentsa It should be noted that not all electron-attracting groups enhance reactivity. The sulfonyl and trifluoro groups, which cannot participate in p* conjugation, retard the rate of SN 2 substitution at an adjacent carbon.

interaction of sp 2 hybridized substitution center with π* LUMO If bond formation at the TS is advanced, resulting in negative charge buildup at carbon, electron withdrawal is more stabilizing. Thus substituents such as carbonyl have their greatest effect on reactions with strong nucleophiles

It should be noted that not all electron-attracting groups enhance reactivity. The sulfonyl and trifluoro groups, which cannot participate in this type of p conjugation, retard the rate of SN 2 substitution at an adjacent carbon. The most important factor is the nature of the p-type orbital that develops at the trigonal bipyramidal carbon in the TS: If the carbon is cationic in character, electron donation from adjacent substituents becomes stabilizing. Adjacent alkoxy substituents act as donors and can stabilize SN 2 TSs that are cationic in character. If bond formation at the TS is advanced, resulting in negative charge buildup at carbon, electron withdrawal is more stabilizing. Thus substituents such as carbonyl have their greatest effect on reactions with strong nucleophiles. Vinyl and phenyl groups can stabilize either type of TS, and allyl and benzyl systems show enhanced reactivity toward both strong and weak nucleophiles. interaction of sp 2 hybridized substitution center with π* LUMO interaction of empty sp 2 orbital with π HOMO interaction of empty sp 2 orbital of benzyl cation with HOMO aromatic π system

5. 9. Relationship between Stereochemistry and Mechanism of Substitution Studies of the stereochemistry are a powerful tool for investigation of nucleophilic substitution reactions. Direct displacement reactions by the SN 2(lim) mechanism are expected to result in complete inversion of configuration. The stereochemical outcome of the ionization mechanism is less predictable, because it depends on whether reaction occurs via an ion pair intermediate or through a completely dissociated ion. Borderline mechanisms may also show variable stereochemistry, depending upon the lifetime of the intermediates and the extent of ion pair recombination.

Scheme 5. 1. Stereochemistry of Nucleophilic Substitution Reactions ionization and internal return partial racemization occurs in aqueous dioxane but that an added nucleophileof (azide ion) results in complete inversion in The observation inversion indicates a concerted the products resulting reaction with both azide ion and mechanism, even in from this weakly nucleophilic solvent. water.

This selection of stereochemical results points out the relative rarity of the idealized SN 1 lim stereochemistry of complete racemization.

5. 9 Relationship between Stereochemistry and Mechanism of Substitution Reaction of alcohols with SOCl 2:

Diazonium Ion Decomposition Water favors In contrast formation to an of ionization a carbocation process with from extensive a neutral racemization, substrate, which whereas initially less polar solvents, generates includinga acetic contactacid, ion pair, lead to deamination more extensive reactions inversion generate as the a cation result that of solvent does participation. not have a closely associated anion. The water molecule formed in the elimination step is captured preferentially from Alcohol Net-retention of for Configuration the front side, Formation: leading to net retention of configuration the alcohol. Ester Formation: Retention and inversion is more similar.

Neighboring Group Participation G: X

5. 10 Neighboring-Group Participation Solvolysis of 2 -acetoxycyclohexyl p-toluenesulfonate: Besides the pronounced difference in rate, the isomeric compounds reveal a striking difference in stereochemistry. The diacetate obtained from the cis isomer is the trans compound (inversion), whereas retention of configuration is observed for the trans isomer. inversion retention

When enantiomerically pure trans-2 -acetoxycyclohexyl tosylate is solvolyzed, the product is racemic trans-diacetate because the acetoxonium intermediate is achiral. evidence for this interpretation ﻭﻗﺘی ﺣﻼﻝ کﺎﻓﺖ ﺩﺭ ﺍﺗﺎﻧﻮﻝ ﺻﻮﺭﺕ ﻣی گیﺮﺩ یﻮﻥ ﺍﺳﺘﻮکﺴﻮﻧیﻮﻡ ﺗﻮﺳﻂ ﺣﻼﻝ ﺑﻪ ﺩﺍﻡ ﻣی ﺍﻓﺘﺪ Racemic mixture

Solvolysis of 4 -chloroalkanol The hydroxy group can act as an intramolecular nucleophile. Table 5. 16. Solvolysis Rates of –Chloro Alcohols

Alkoxy group is a weak nucleophile, but it can function as a neighboring nucleophile. Ex: solvolysis of the isomeric p-bromobenzenesulfonate esters A and B leads to identical product mixtures, indicating the involvement of a common intermediate a cyclic oxonium ion.

Transanular participation of ether oxygen

In general, any system that has a nucleophilic substituent situated properly for back-side displacement of a leaving group at another carbon atom of the molecule can be expected to display neighboringgroup participation. The electrons of carbon-carbon double bonds can also become involved in nucleophilic substitution reactions. This participation can facilitate the ionization step if it leads to a carbocation having special stability

p Electron of C=C Participation Acetolysis of Norbornenes The reaction product in the case of syn somer is derived from a rearranged carbocation that is stabilized by virtue of being allylic

Extent of p Electron of C=C Participation Substitution at C-7 position of Norbornenes
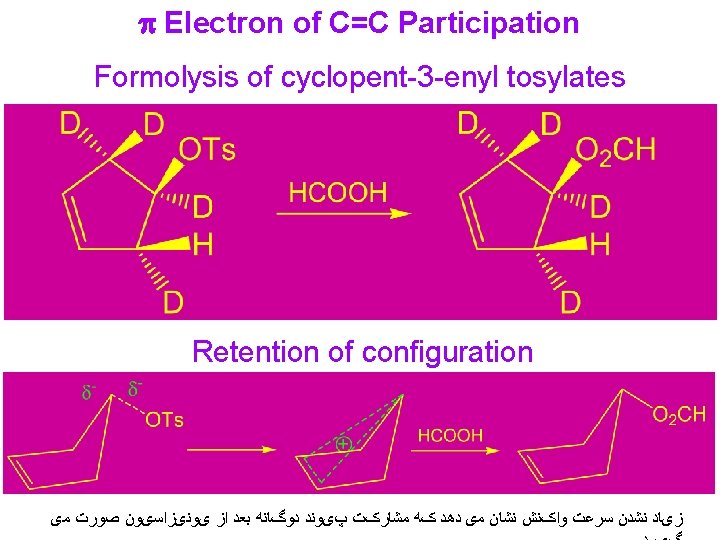

Synthetic Use of p Electron of C=C Participation Acetolysis of 2 -cyclopent-3 -enylethyl tosylate: Participation of carbon-carbon double bonds in solvolysis reactions is revealed in some cases by isolation of products with new carbon-carbon bonds.
 b-phenyl group participation Evidence for this type of")
Aromatic p Electron Participation (Phenonium Ion) b-phenyl group participation Evidence for this type of participation was first obtained by a study of the stereochemistry of solvolysis of 3 -phenyl-2 -butyl tosylates bridged carbocation enantio-pure erythro prduct

Isotope labeling: The extent participation can be quantitatively measured by comparing The of relative importance of aryl participation is a function of the ratesubstituents of direct displacement, on the ring. ks , with the rate of aryl-assisted solvolysis, designated k. D. The extent of label scrambling increases as solvent nucleophilicity decreases. In systems with EWG substituents, the aryl ring does not participate effectively and only the process described by ks contributes to the rate. r = -0. 7 characteristic of a Weak substituent effect Positively charged T. S.


5. 11 Rearrangement of Carbocations 1, 2 -H-Shift or 1, 2 -alkyl-Shift : Driving Force : Formation of more stable carbocation
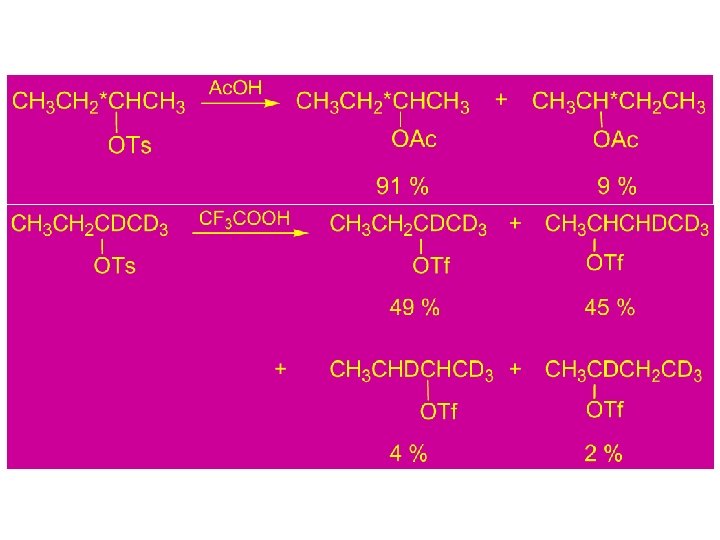
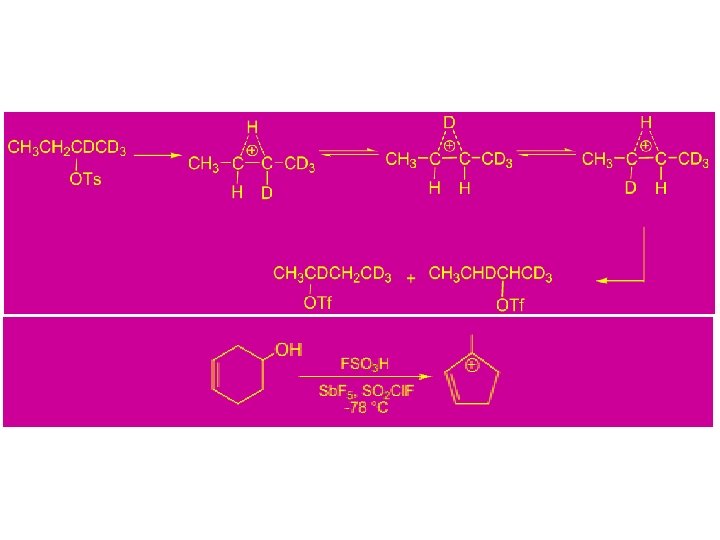
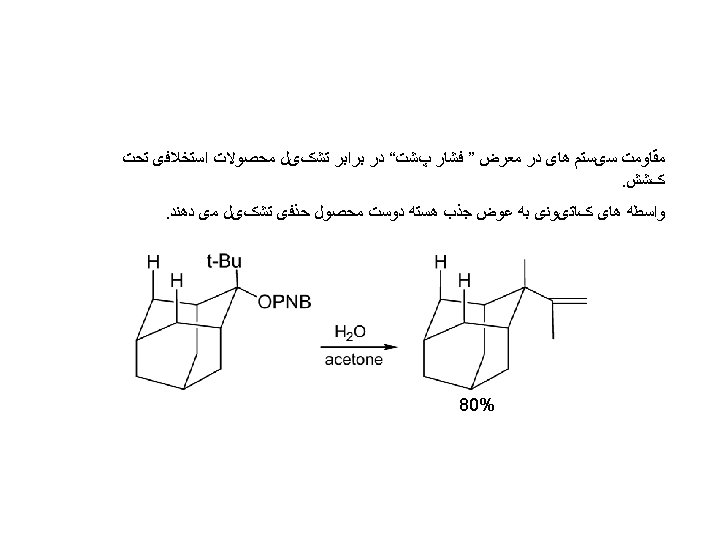
 In acetolysis, a large part of the reaction must be occurring via")
Conclusion: 1) In acetolysis, a large part of the reaction must be occurring via direct nucleophilic participation by the solvent or rapid ion pair capture so that only a relatively small amount of hydride shift occurs. 2) In non-nucleophilic super acid media, the cations are relatively long-lived and undergo several rearrangements, eventually leading to the most stable accessible ion.

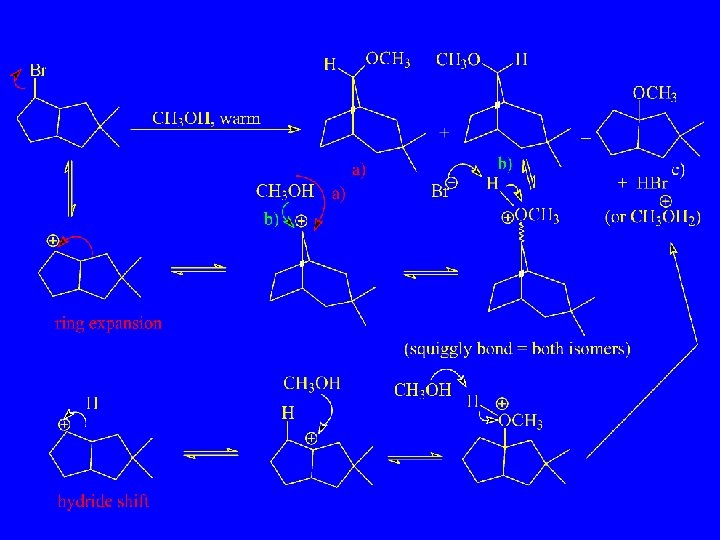
H-Shifts between carbon atoms separated by several atoms 1, 5 -H- Shifts: Cyclononyl-1 -14 C tosylate

Reaction of cyclooctene with CF 3 COOD

Hydride-bridge ion in which the bridging hydride is located in a bicyclic cage. Ring Contraction
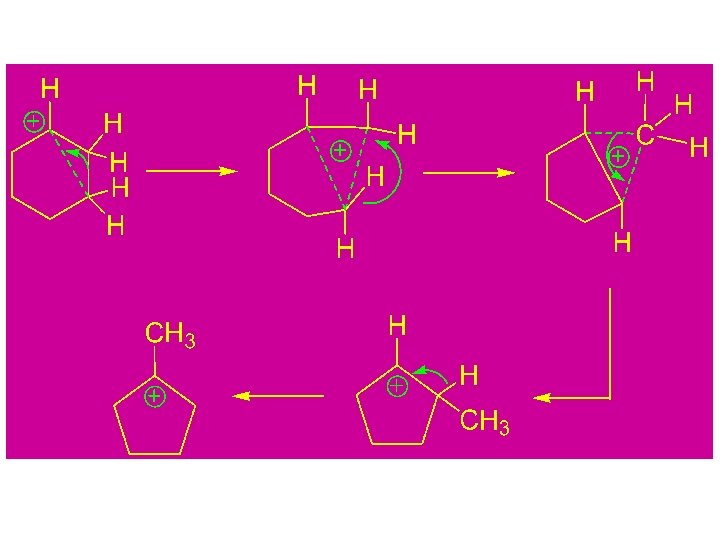

Carbocation Rearrangement Facilitation via The Product Stabilization by a Functional Group Pinacol rearrangements

Study of Carbocation Rearrangements by NMR at low Temperature in Super Acid Media

• In the discussion of carbocation rearrangements, we encountered examples of bridged ions that require expansion of bonding concepts beyond the two-center, two-electron bonds that suffice for most stable organic molecules. • These bridged carbocations, involve delocalization of electrons and formation of three-center, twoelectron bonds, and are sometimes called nonclassical ions.

5. 12 The Norbornyl Cation and Non-Classical Carbocations Optically Active Exo Brosylate → Exo Product (100% racemization) Optically Active Endo Brosylate → Exo Product (98% racemization) Back
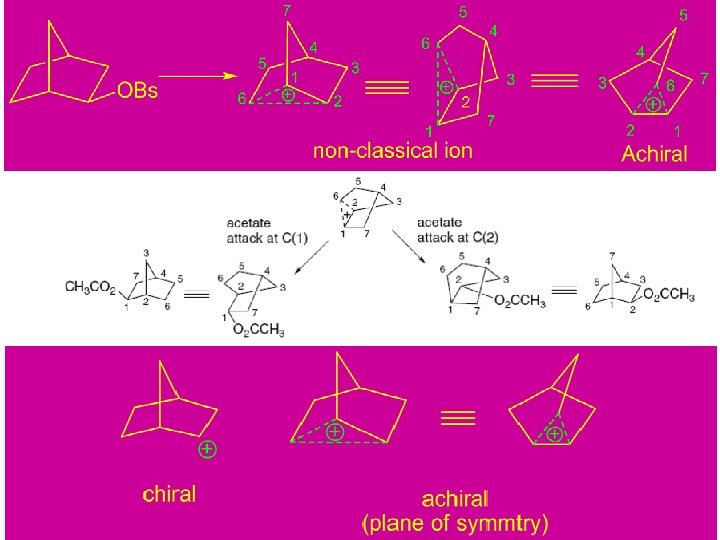
![Bicyclo[3. 2. 1]octyl acetate](http://slidetodoc.com/presentation_image_h/91c922700c45a9aef17b6630d67737a5/image-111.jpg "Bicyclo[3. 2. 1]octyl acetate")
Bicyclo[3. 2. 1]octyl acetate

The description of the nonclassical norbornyl cation developed by Winstein implied that the bridged ion is stabilized relative to a secondary ion by C−C bond delocalization. H. C. Brown: Rapidly equilibrating classical ion and non-classical ion as T. S.
")
Fig. 5. 7. Contrasting energy profiles for stable and unstable bridged norbornyl cation. (A) Bridged ion is a transition structure for rearrangement between classical structures. (B) Bridged ion is an intermediate in rearrangement of one classical structure to the other. (C) Bridged ion is the most stable structure.

Solvolysis exo and endo 2 -phenyl-norbornyl-pnitrobenzoate in aqueous Dioxane
 NMR Spectroscopy in non-nucleophilic media (supper acids). The cation")
Evidences for Non-Classical Ions: 1) NMR Spectroscopy in non-nucleophilic media (supper acids). The cation was generated in Sb. F 5 -SO 2 -SOF 2 and the temperature dependence of the proton magnetic resonance spectrum was examined. 2) X-Ray crystallography

Provide a Mechanism
- Slides: 117