Chapter 3 Structural Effects on Stability and Reactivity

Chapter 3 Structural Effects on Stability and Reactivity learning tools to use deciphering reaction mechanisms -> a mechanism can never really be proven. Energy surfaces and related concepts Energy surface

Reaction coordinate diagrams Reaction coordinate: the minimum energy pathways, or the pathway we depict as the weighted average of all the pathways Reaction coordinate diagram

Intermediates: any chemical structures that last longer than the time for a typical bond vibration (10 -13 to 10 -14 s) Rate-determining step: the step with the highest barrier. Activation energy
: a proportionality constant between concentration of reactants")
Rates and rate constants Rate constant (k): a proportionality constant between concentration of reactants and reaction rate
: a proportionality constant between concentration of")
Reaction order and rate laws Rate constant (k): a proportionality constant between concentration of reactants and reaction rate Rate law a + b + c = reaction order -> determined by experimental measurements of the rates of reaction: does not give any information about mechanism Molecularity: the number of molecules involved in the transition state of the reaction Elementary reaction: single-step reaction (단일반응) Unimolecular: only single molecule is involved in the transition state, eg) Cope rearrangement Bimolecular: two molecules are involved in the transition state, eg) SN 2 Termolecular: three molecules are involved in the transition state Molecularity applies only to elementary reactions and is basically a statement about the mechanism of the reaction.

Transition state theory and related topics The mathematics of transition state theory: pre-equilibrium between the reactants and activated complex To understand the nature of the rate constant k -> analyze the energetic and entropic components of reaction process + + k + + → [AB ] A+B← → C + AB + rate = + [AB+] + K+ = [A][B] Equilibrium constant of the formation of activated complex A+B + + = k K [A][B] + + + + DG = DH - TDS = -RTn. K + kk. BT + + -DG /RT e h = k [A][B] rate = k= kk. BT h e + + -DG /RT [A][B] + + + D G /RT + K =e k: transmission coefficient ~ 1 k. B: Boltzmann constant h: Planck’s constant T: absolute temperature C

kk. BT + + -DG /RT e h kk. BT -DH++/RT DS++/R e = e h kk. B DS++/R -DH++/RT e = e T h k= + + -DH /RT T = C e Eyring equation C= kk. B h + + e. DS /R Relationship to the Arrhenius rate law A: frequency factor (pre-exponential factor) Ea: activation energy T: absolute temperature Eyring equation: analyzes a microscopic rate constant for a single-step conversion of a reactant to a product. In a multistep process involving reactive intermediates, there is an Eyring equation and thus a DG‡ for each and every step. Arrhenius rate law: arises from empirical observations of the macroscopic rate constants for a particular conversion, such as A going to B by various paths. It is ignorant of any mechanistic considerations, such as whether one or more reactive intermediates are involved in the overall conversion of A to B. Ea describes the overall transformation.

Boltzmann distributions and temperature dependence The higher temperature reaction proceeds faster due to the larger area under the curve past the activation energy Boltzmann distribution

Experimental determinations of activation parameters and Arrhenius parameters Eyring equation Arrhenius equation T -> k --- DH‡, DS‡, DG‡


Examples of activation parameters and their interpretations Diels-Alder reaction Gas phase DH‡ = 15. 5 kcal/mol DS‡ = -34 eu Gas phase DH‡ = 52 kcal/mol DS‡ = 19 eu DH‡ : the latter > the former why? the former; concerted reaction -> bond making accompanies bond breaking DS‡ : the latter > the former why? The former -> two molecules become one molecule -> the loss of translational and rotational degrees of freedom the latter -> one molecule becomes three molecules Claisen rearrangement DS‡ = -8 eu

1, 2, 4, 6 -> molecules combine 3, 5, 7 -> bond breaking 1; -26 eu -> 26 cal/mol K x 298 K = 7. 75 kcal/mol in DG‡ -> decrease in the rate constant by a factor of 500, 000, relative to a reaction with a DS‡ = 0

Postulates and principles related to kinetic analysis Hammond postulate Transition states are transient in nature and generally cannot be directly characterized by experimental means. This postulate is likely the most widely used principle for estimating the structures of activated complexes that give us insights into chemical reactivity. ; the activated complex most resembles the adjacent reactant, intermediate, or product that it is closest in energy to. exothermic endothermic

The Hammond postulate does not predict the height of the barrier compared to the reactant and product. To obey the Hammond postulate 이런 경우도 있다. 그러나 대부분의 경우 7. 9와 같은 reaction coordinates를 갖는다

The reactivity vs. selectivity principle The more reactive a compound is, the less selective it will be. More reactive molecules -> being higher in energy or having more exothermic reactions. -> the more reactive species will produce a transition state that more resembles the reactant. Thus, the transition state is not very sensitive to the structure of other components involved in the reaction, and it is affected little by the structure of the product. -> If the reaction is not sensitive to the structure of the product, it can not select between different products and hence is not selective.
. -> H·")
Bond strength Cl· is much more reactive than Br· (H-Cl > H-Br). -> H· abstraction by Cl· is an exothermic reaction -> very little radical character on carbon in TS -> quite indiscriminate about which H is abstracted (chlorination is much less selective than bromination) H· abstraction by Br· is an endothermic reaction -> large radical character on carbon in TS -> H· abstraction by Br· is quite selective

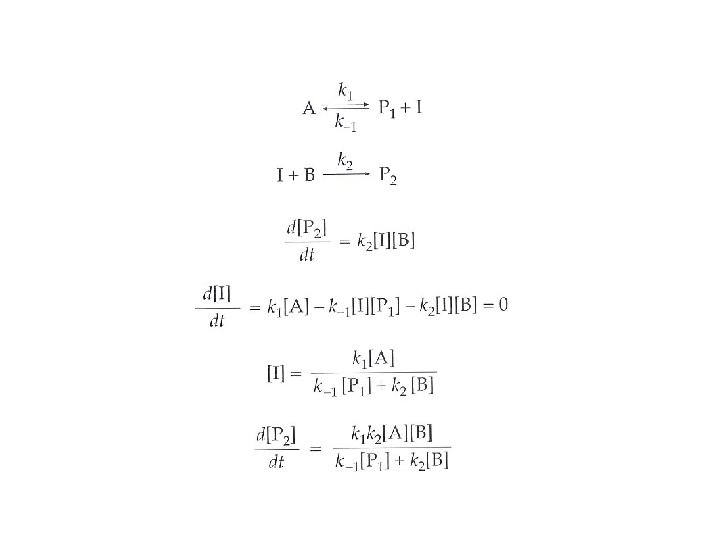
The Curtin-Hammett principle This principle states that the ratio of products is determined by the relative heights of the highest energy barriers leading to the different products, and is not significantly influenced by the relative energies of any isomers, conformers, or intermediates formed prior to the highest energy transition states. This principle is applicable when the barriers interconverting the different starting points is much lower than the barriers to form products. Rapid equilibrium → P 1 ← I 1 I 2 → P 2 ← Keq Product ratio = [P 1]/[P 2] = (d[P 1]/dt) / (d[P 2]/dt) = ka[I 1]/kb[I 2] = (ka / kb) x (1/Keq) kk. BT e + + -DG 1 /RT h x e –(-DGo /RT) + kk. BT –DG 2+/RT e h + + ++ DG 2 ++ DGo)/RT (D G 1 = e = + + ka DG 1 + DG 2 + kb DGo + + DG 1+, DG 2+>> DGo very rapid equilibrium = e + + + (-DG 1 ++ DG 2 )/RT

Kinetic vs. thermodynamic control Thermodynamic control: product composition is governed by equilibrium of the system. Kinetic control: product composition is governed by reaction rate or activation energy.
 Li+ -N(i. Pr)2 -> strong, sterically hindered base")
LDA Na. OH LDA (lithium diisopropylamide) Li+ -N(i. Pr)2 -> strong, sterically hindered base in tetrahydrofuran (THF) -> kinetic enolates Na. OH in H 2 O or Et. OH Weak base, thermodynamic control -> thermodynamic enolates

Kinetic experiments Kinetic analyses for simple mechanisms Goal of kinetics: to establish a quantitative relationship between the concentrations of reactants and / or products, and the rate of the reactions. First order kinetics A -> P half-life time (t 1/2): time required for 50% of the starting material to be consumed v= [A]0/2 -> t 1/2 =ln 2/k = 0. 693/k ln[A] slope = -k (1/sec) t [A]0 = [A] + [P] - plot of ln[A] vs t will be linear - t 1/2 is constant - k should not depend on [A]0

ii가 rds에 관여하지 않는다. -> Support a rate-determining unimolecular decomposition of i to give cyclopentyne rds Bimolecular reaction
![Second order kinetics A + B -> P [A]0 = [A] + [P] [B]0](http://slidetodoc.com/presentation_image_h2/8646e07ca79dd4fc5afb5772048cd983/image-22.jpg "Second order kinetics A + B -> P [A]0 = [A] + [P] [B]0")
Second order kinetics A + B -> P [A]0 = [A] + [P] [B]0 = [B] + [P] [A]- [A]0 = [B] - [B]0 = k[A]([B]0 - [A]0 +[A]) = -d[A]/dt 1 [B]0 - [A]0 ln [A]0([B]0 - [A]0 +[A]) [B]0[A] = kt

no SN 2 reaction

Pseudo-first order kinetics Second order kinetics: too complicating -> Pseudo-first order kinetics A + B -> P B -> a large excess (usually 10 equivalents or more) -> [B] will change little during the course of the reaction : [B] ~ [B]0 -> pseudo-first order kinetics k’ = kobs = k[B]0 ln[A] kobst slope = -kobs = -k[B]0 t Determination of k Vary concentration of the excess [B] kobs slope = k [B]

Initial rate kinetics - In case that the reaction is slow, it is difficult to follow the reaction to several half-lives in order to obtain a reliable rate constant - Many reactions start to have significant competing pathways as the reaction proceeds, causing deviations from the ideal behaviors. Initial rate kinetics: only follow the reaction to 5% or 10% completion ([A] ~ [A]0), thereby avoiding complications that may arise later in the reaction and/or allowing us to solve for rate constants in a reasonable time period. -> this approach is inherently less accurate than a full monitoring of a reaction over several half-lives, but often it is the best we can do. = k[A]0 [P] = k[A]0 t Plotting [P] versus t over the first few percent of the reaction gives a line whose slope is k[A]0 slope = k[A]0 k


Complex reactions – deciphering mechanisms Steady state kinetics Most organic, bioorganic, and organometallic mechanisms have more than one step, and frequently involve reactive intermediates. -> such mechanisms can produce complex rate laws that are difficult to analyze. The concentration of the reactive intermediate is constant during the reaction since the reactive intermediate (short life-time) is not accumulated during the reaction Steady state approximation (SSA) A + B -> P Rate of the formation of I = rate of disappearance of I

Enzyme kinetics: steady state kinetics k 1 E kcat E + S ← → E·S → P k-1 + → ← → + S E E·S: Michaelis complex P E·S rate = d[P]/dt = kcat[ES] d[ES]/dt = k 1[E][S] – k-1[ES] - kcat[ES] = 0 [E]0 = [E] + [ES] [E] = [E]0 - [ES] k 1([E]0 -[ES])[S] – k-1[ES] - kcat[ES] = 0 [ES] = [E]0[S] Km + [S] Km = (k-1 + kcat)/k 1 rate = d[P]/dt = kcat[ES] = kcat[E]0[S] Km + [S] Michaelis-Menten equation Kinetic parameters to be determined for enzymatic reactions: kcat and Km
![각 substrate 농도에 따른 initial rate 측정 (실험) 계산 Vmax rate = kcat[E]0[S] Km](http://slidetodoc.com/presentation_image_h2/8646e07ca79dd4fc5afb5772048cd983/image-29.jpg "각 substrate 농도에 따른 initial rate 측정 (실험) 계산 Vmax rate = kcat[E]0[S] Km")
각 substrate 농도에 따른 initial rate 측정 (실험) 계산 Vmax rate = kcat[E]0[S] Km + [S] 1/rate = 1/Vmax + Km/(Vmax [S]) Vmax = kcat[E]0 Km << [S] 1/rate 1. kcat: catalytic constant turnover number: the maximum number of substrate molecules converted to products per active site per unit time 2. Km : apparent dissociation constant that may be treated as the overall dissociation constants of all enzyme-bound species. slope = Km/Vmax 1/[S] Lineweaver-Burk plot



B에 대하여 zero order kinetics

Methods for following kinetics Reactions with half-lives greater than a few seconds No special technique requirement for generating the reactants and/or mixing the reactants -> the kinetic analysis can be performed with chromatographic and spectroscopic methods. Chromatographic analysis : HPLC or GC Spectroscopic analysis: continuous analysis is possible (UV, fluorescence) or NMR Fast kinetics Half-life; a few seconds or less -> Special technique requirement for generating the reactants and/or mixing the reactants. Stopped flow analysis Flash photolysis Pulse radiolysis

Studies of reaction mechanisms Isotope effects: important for mechanistic studies k m 1 m 2 Hooke’s law v = 1/(2 p) k/ k = force constant = reduced mass, m 1 m 2/(m 1 +m 2) E = hv = hc/l = hcv v = wavenumber (cm-1) v. CH k. CH/ CH CD v. CH = = = v. CD k. CD/ CD CH k. CD ~ k. CH v. CD = = 24/14 12/13 v. CH = 3000/1. 36 ~ 2200 cm-1 (IR spectra) 1. 36 = 1. 36
 v. CH")
Vibrational energy level e 0 = hv/2 : zero point energy (ZPE) v. CH v. CD = 1. 36 e 0, CH = 1. 36 e 0, CD e 0, CH = 18 k. J/mol e 0, CD = 13 k. J/mol

C···H or C···D e 0, CH = 18 k. J/mol e 0, CD = 13 k. J/mol ZPECH, TS = ZPECD, TS k. H k. D In general, Maximum value ‡ ‡ = e(-DG H +DGD )/RT = e 5000 j/RT = e 2. 00 = 7. 4 at 25 o. C 20. 2 at -73 o. C 1026 at -263 o. C k. H k. D <7. 5

Primary KIE in case that a bond to the isotopically substituted atom is broken in the ratedetermining step k. H k. D = 6. 1 (25 o. C) -> deprotonation is rds If the second step is rds, what happens? Answer) k. H =1 k. D k. H k. D = 4. 6 (77 o. C) Product-like TS; large radical character (largely broken) -> large KIE k. H k. D = 1. 5 (77 o. C) Reactant-like TS; small radical character (a little broken) -> small KIE


Secondary KIE in case that substituted hydrogen atom is not directly involved in the reaction k. H k. D = 0. 7 ~ 1. 5 k. H k. D > 1; normal KIE k. H k. D < 1; inverse KIE r. C-D < r. C-H PE r. C-D r. C-H Distance (r)
 k. H k. D bond length;")
a-effect sp 3 -> sp 2 (transition state) k. H k. D bond length; sp 3 > sp 2 k. H k. D = 1. 37 (50 o. C) > 1 (normal KIE) sp 2 -> sp 3 (transition state) k. H k. D bond length; sp 3 > sp 2 k. H k. D = 0. 73 (25 o. C) < 1 (inverse KIE)

sp 2 Pathway A: nucleophilic addition to the flavin sp 2 -> sp 3 ; expected, inverse KIE Pathway B: an electron transfer to the flavin sp 2 -> ~sp 2 ; expected, little KIE D-amino acid oxidase k. H k. D = 0. 84 large inverse KIE Support pathway A

b-effect k. H k. D = 1. 31 Why? hyperconjugation Bond energy; EC-D > EC-H -> the contribution of C-D bond hyperconjugation is smaller than that of C-H. 1 2 H 2 O carbocation solvolysis k. H k. D = 1. 14 per D 1 -> product k. H k. D Orthogonal = 0. 99 no KIE (no orbital overlap) 2

Long range KIE k. H k. D = 0. 86 r. C-D < r. C-H Size: CD 3 < CH 3 Size: C(CD)3 < C(CH)3 k. H k. D = 1. 107 Steric hindrance is released by going to TS

Tunneling Sometimes KIE of 50 or larger are observed for comparisons of H vs D. -> Tunneling is a quantum mechanical phenomenon involving penetration of the wavefunction for the molecule through the barrier for the reaction than over it. -> the tunneling is very sensitive to the mass of the tunneling particle. -> tunneling is usually associated with proton or hydrogen transfers or electron transfers.

k. H k. D = 74

Substituent effects The origin of substituent effects Field effects - is one of that originates from such a through-space interaction - is often very hard to separate from inductive effects A quaternary ammonium with a full positive charge produces an electric field that can influence distant atoms in the same or neighboring molecules.

Inductive effects - results from the ability of an atom or group of atoms to withdraw or donate electrons through bond. F; poor at making HBs; but more stable collagen; inductive effects

Resonance effects - the ability of an atom or group of atoms to withdraw or donate electrons through p bonds. Polarizability effects - defined as the extent to which the electron cloud of the structure can undergo distortion. Steric effects Solvation effects Field effects, Inductive effects, Resonance effects, Polarizability effects: electronic effects
- Slides: 49