Cdric Notredame Comparative Bioinformatics Group Bioinformatics and Genomics

最佳的多重序列比對方法針對基因組 領域 Cédric Notredame Comparative Bioinformatics Group Bioinformatics and Genomics Program

Getting the best out of multiple sequence alignment methods in the genomic era Cédric Notredame Comparative Bioinformatics Group Bioinformatics and Genomics Program

Which Tool for Which Sequence ?

In- Sil. Vo Biology l In Silico Biology – l In Vivo Biology – l Making Sense of digital data Recording data in a living Cell In Sil. Vo Biology – Connect In-Vitro and In-Vivo l l In-Vivo: High-throughput recording In-Silico: High-Throughput analysis

Is it Possible to Compare all Types of Sequences ? l Non Transcribed World – Genes/Full Genomes l – Promoter Regions l l – Meta-Aligner Motifs Finders Nucleosome l l Lagan, TBA ? ? ? Multiple Genome Aligners – – – Not Very Accurate Very Fast Deal with rearrangements

Multiple Genome Alignments and re-sequencing l Before – – l Re-sequence Human Genomes Map the Reads onto the reference genome Now – – Re-sequence Assemble Align Non trivial with very large datasets

Is it Possible to Compare all Types of Sequences ? l RNA Comparison – – l Less Accurate than Proteins Secondary Structures nc. RNA World – Sankoff l l – – Time O(L 2 n) Space O(L 3 n) Consan R-Coffee

Is it Possible to Compare all Types of Sequences ? l Protein Comparisons – – l Very Accurate 3 D-Structure Improves it Protein Aligners – – – Clustal. W T-Coffee 3 D-Coffee

What Changes with 1000 Genomes?

Phylogeny Vs Function l Function – – l Low level => Biochemistry => Protein Domains High Level => Metabolic Pathway => Orthology – – Phylogenetic Analysis =>Accurate Alignments

one 2 many apparent one 2 one many 2 many one 2 one Duplication node Speciation node or leaf (Adpated from “Going beyond AGC and T, E. Birney)

Using The tree Correct Tree Correct Orthologous Assignment Correct Functional Prediction

The Alignment that Hides The Forest…

Phylogenetic Trees and Multiple Sequence Alignments

Phylogenetic Trees and Multiple Sequence Alignments

Genomic Era: The Goal l 10. 000 Sequences: interspecies 1 Billion: Re-sequencing l Incorporation of ALL experimental Data l – l Alignments suitable for all applications of comparative genomics – – l Structure, Genomic, Ch. Ip-Chip, Ch. Ip-Seq… Homology Modeling (function) Functional Analysis Phylogenetic Reconstruction 3 D-Modelling Accurate Alignments for ALL kind of data l l l Non Transcribed DNA Translated DNA

Genomic Era Challenges l Accuracy – – l Proteins: 30% is the limit DNA/RNA 70% is the limit Data Integration – – l – Scale – With too many sequences algorithms lose in accuracy l Structure Homology Genomic Structure Function Proteomics Methods – – Wealth of alternative methods Poorly Characterized

Consistency and Data Integration l Most methods rely on the progressive algorithm l Consistency based methods have been designed as an extension l Consistency based alignment methods have been designed to: – – – Better extract the signal contained in the data Integrate/Confront existing methods Integrate/Confront heterogeneous types of Information

The Progressive Alignment Algorithm

T-Coffee and Concistency… Seq. A GARFIELD THE LAST FAT CAT Seq. B GARFIELD THE FAST CAT Seq. C GARFIELD THE VERY FAST CAT Seq. D THE FAT CAT Seq. A Seq. B Seq. C Seq. D GARFIELD ---- THE THE LAST FAST VERY ---- FA-T CA-T FAST FA-T CAT --CAT

T-Coffee and Concistency… Seq. A GARFIELD THE LAST FAT CAT Seq. B GARFIELD THE FAST CAT --- Prim. Weight =88 Seq. A GARFIELD THE LAST FA-T CAT Seq. C GARFIELD THE VERY FAST CAT Prim. Weight =77 Seq. A GARFIELD THE LAST FAT CAT Seq. D ---- THE ---- FAT CAT Prim. Weight =100 Seq. B GARFIELD THE ---- FAST CAT Seq. C GARFIELD THE VERY FAST CAT Prim. Weight =100 Seq. C GARFIELD THE VERY FAST CAT Seq. D ---- THE ---- FA-T CAT Prim. Weight =100

T-Coffee and Concistency… Seq. A GARFIELD THE LAST FAT CAT Seq. B GARFIELD THE FAST CAT --- Prim. Weight =88 Seq. A GARFIELD THE LAST FA-T CAT Seq. C GARFIELD THE VERY FAST CAT Prim. Weight =77 Seq. A GARFIELD THE LAST FAT CAT Seq. D ---- THE ---- FAT CAT Prim. Weight =100 Seq. B GARFIELD THE ---- FAST CAT Seq. C GARFIELD THE VERY FAST CAT Prim. Weight =100 Seq. C GARFIELD THE VERY FAST CAT Seq. D ---- THE ---- FA-T CAT Prim. Weight =100 Seq. A GARFIELD THE LAST FAT CAT Seq. B GARFIELD THE FAST CAT --- Weight =88 Seq. A GARFIELD THE LAST FA-T CAT Seq. C GARFIELD THE VERY FAST CAT Seq. B GARFIELD THE ---- FAST CAT Weight =77 Seq. A GARFIELD THE LAST FA-T CAT Seq. D ---- THE ---- FA-T CAT Seq. B GARFIELD THE ---- FAST CAT Weight =100

T-Coffee and Concistency… Seq. A GARFIELD THE LAST FAT CAT Seq. B GARFIELD THE FAST CAT --- Weight =88 Seq. A GARFIELD THE LAST FA-T CAT Seq. C GARFIELD THE VERY FAST CAT Seq. B GARFIELD THE ---- FAST CAT Weight =77 Seq. A GARFIELD THE LAST FA-T CAT Seq. D ---- THE ---- FA-T CAT Seq. B GARFIELD THE ---- FAST CAT Weight =100

T-Coffee and Concistency…

T-Coffee and Concistency…

Methods Scalability Data

A Brief History of Consistency A Long Chain of Small Contributions…
 – l Iterative strategy using consistency AMAP (Schwarz,")
Consistency Based Algorithms l Gotoh (1990) – l Iterative strategy using consistency AMAP (Schwarz, 2007) – – l – Martin Vingron (1991) – – Dot Matrices Multiplications Accurate but too stringeant l FSA ( Patcher, 2009) – l Dialign (1996, Morgenstern) – – l T-Coffee (2000, Notredame) – – l Concistency Agglomerative Assembly Concistency Progressive algorithm Prob. Cons (2004, Do) – – T-Coffee with a Bayesian Treatment Biphasic Gap Penalty Prob. Consistency Replace Progressive alignment with simulated Annealing Hard to distinguish from Prob. Cons – AMAP with automated parameter estimation Hard to distinguish from Prob. Cons

Choosing the right modeling method M-Coffee

Combining Many MSAs into ONE Clustal. W MAFFT T-Coffee MUSCLE ? ? ? ?

Consistency and Accuracy

Integrating New Types of Data Template Based Sequence Alignments

Templates TARGET Template Aligner TARGET Experimental Data … Template Alignment Template-Sequence Alignment Template based Alignment of the Sequences Primary Library

Exploring The Template World Template Generator Alignment Method RNA Structure Prediction RNA Aligner Protein Structure BLAST vs PDB 3 D Aligner Profile BLAST vs NR Profile/Profile Alignment Gene Structure ENSEMBL Genome Aligner Promoter Transfac Meta-Aligner

Exploring The Template World Template Generator Alignment Method Mode RNA Structure Prediction RNA Aligner R-Coffee Protein Structure BLAST /PDB 3 D Aligner 3 D-Coffee Profile BLAST/NR Profile/Profile PSI-Coffee Gene Structure ENSEMBL Genome Aligner Exoset Promoter Transfac Meta-Aligner Meta-Coffee

3 D-Coffee/Expresso Incorporating Structural Information

Expresso: Finding the Right Structure Sources BLAST SAP Templates Template Alignment Source Template Alignment Remove Templates Library

PSI-Coffee Homology Extension

Exploring The Template World

What is Homology Extension ? -Simple scoring schemes result in alignment ambiguities L ? L L

What is Homology Extension ? L L L Profile 1 L L L I V I L L L L Profile 2

What is Homology Extension ? L L L I V I L L L L Profile 1 Profile 2

PSI-Coffee: Homology Extension Sources BLAST Profile Aligner Templates Template Alignment Source Template Alignment Remove Templates Library

Benchmarks

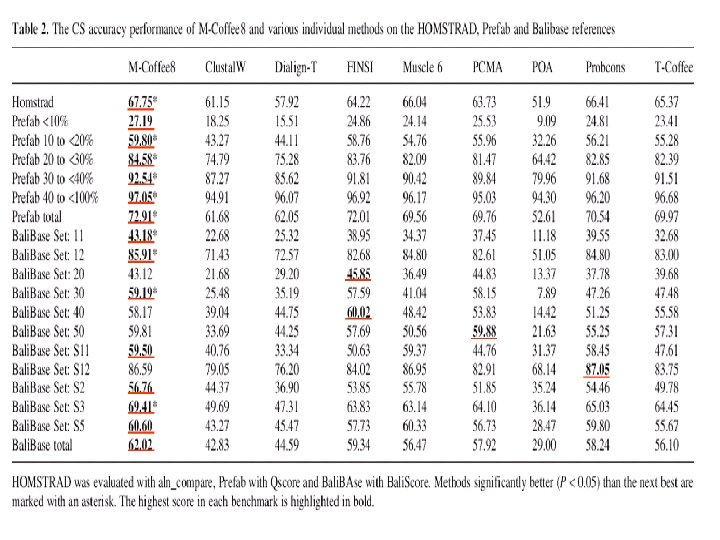
Method Template Score Clustal. W-2 Progressive NO 22. 74 PRANK Gap NO 26. 18 MAFFT Iterative NO 26. 18 Muscle Iterative NO 31. 37 Prob. Consistency NO 40. 80 Prob. Cons Mono. Phasic NO 37. 53 T-Coffee Consistency NO 42. 30 M-Coffe 4 Consistency NO 43. 60 PSI-Coffee Consistency Profile 53. 71 PROMAL Consistency Profile 55. 08 PROMAL-3 D Consistency PDB 57. 60 3 D-Coffee Consistency PDB 61. 00 Comment Science 2008 Expresso Score: fraction of correct columns when compared with a structure based reference (BB 11 of Bali. Base).

Method Template Score Clustal. W-2 Progressive NO 22. 74 PRANK Gap NO 26. 18 MAFFT Iterative NO 26. 18 Muscle Iterative NO 31. 37 Prob. Consistency NO 40. 80 Prob. Cons Mono. Phasic NO 37. 53 T-Coffee Consistency NO 42. 30 M-Coffe 4 Consistency NO 43. 60 PSI-Coffee Consistency Profile 53. 71 PROMAL Consistency Profile 55. 08 PROMAL-3 D Consistency PDB 57. 60 3 D-Coffee Consistency PDB 61. 00 Comment Science 2008 Consistency Expresso Score: fraction of correct columns when compared with a structure based reference (BB 11 of Bali. Base).

Method Template Score Clustal. W-2 Progressive NO 22. 74 PRANK Gap NO 26. 18 MAFFT Iterative NO 26. 18 Muscle Iterative NO 31. 37 Prob. Consistency NO 40. 80 Prob. Cons Mono. Phasic NO 37. 53 T-Coffee Consistency NO 42. 30 M-Coffe 4 Consistency NO 43. 60 PSI-Coffee Consistency Profile 53. 71 PROMAL Consistency Profile 55. 08 PROMAL-3 D Consistency PDB 57. 60 3 D-Coffee Consistency PDB 61. 00 Comment Science 2008 Homology Extension Expresso Score: fraction of correct columns when compared with a structure based reference (BB 11 of Bali. Base).

Method Template Score Clustal. W-2 Progressive NO 22. 74 PRANK Gap NO 26. 18 MAFFT Iterative NO 26. 18 Muscle Iterative NO 31. 37 Prob. Consistency NO 40. 80 Prob. Cons Mono. Phasic NO 37. 53 T-Coffee Consistency NO 42. 30 M-Coffe 4 Consistency NO 43. 60 PSI-Coffee Consistency Profile 53. 71 PROMAL Consistency Profile 55. 08 PROMAL-3 D Consistency PDB 57. 60 3 D-Coffee Consistency PDB 61. 00 Comment Science 2008 Structural Extension Expresso Score: fraction of correct columns when compared with a structure based reference (BB 11 of Bali. Base).

T-Coffee and The World -Some Templates are obtained with a BLAST -Queries can be sent to the EBI or the NCBI -No Need for a Local BLAST installation BLAST/ SOAP Users sequences

Incorporating RNA Information Within the T-Coffee Algorithm

nc. RNAs Can Evolve Rapidly A A C CA C G G A A CG G G C A T C G A A C CA C G G A A CG G C G T A CCAGGCAAGACGGGACGAGAGTTGCCTGG T A G C CCTCCGTTCAGAGGTGCATAGAACGGAGG C G **-------*--**---*-**------** C G T A C G
 CC--AGGCAAGACGGGACGAGAGTTGCCTGG CCTCCGTTCAGAGGTGCATAGAAC--GGAGG")
nc. RNAs Can Evolve Rapidly CCAGGCAAGACGGGACGAGAGTTGCCTGG CCTCCGTTCAGAGGTGCATAGAACGGAGG **-------*--**---*-**------** Sequence Alignment (Maximizing Identity) CC--AGGCAAGACGGGACGAGAGTTGCCTGG CCTCCGTTCAGAGGTGCATAGAAC--GGAGG ** * * *** ** -Incorrect -Not Predictive

The Holy Grail of RNA Comparison: Sankoff’ Algorithm

R-Coffee Extension TC Library C C G G Score X C C Score Y C C l l G G Goal: Embedding RNA Structures Within The T-Coffee Libraries The R-extension can be added on the top of any existing method.

R-Coffee + Structural Aligners Method Avg Braliscore Net Improv. direct +T +R -----------------------------Stemloc 0. 62 0. 75 0. 76 104 113 Mlocarna 0. 66 0. 69 0. 71 101 133 Murlet 0. 73 0. 70 0. 72 -132 -73 Pmcomp 0. 73 142 145 T-Lara 0. 74 0. 69 -36 -8 Foldalign 0. 75 0. 77 72 73 -----------------------------Dyalign --0. 63 0. 62 ----Consan --0. 79 -------------------------------Improvement= # R-Coffee wins - # R-Coffee looses over 170 test sets

R-Coffee + Regular Aligners Method Avg Braliscore Net Improv. direct +T +R -----------------------------Poa 0. 62 0. 65 0. 70 48 154 Pcma 0. 62 0. 64 0. 67 34 120 Prrn 0. 64 0. 61 0. 66 -63 45 Clustal. W 0. 65 0. 69 -7 83 Mafft_fftnts 0. 68 0. 72 17 68 Prob. Cons. RNA 0. 69 0. 67 0. 71 -49 39 Muscle 0. 69 0. 73 -17 42 Mafft_ginsi 0. 70 0. 68 0. 72 -49 39 ------------------------------ Improvement= # R-Coffee wins - # R-Coffee looses over 388 test sets
 Template Based Alignments R-Coffee Scaled Consistency Meta-Methods")
Genomic Era Challenges Conclusion Homology Extension (Proteins) Template Based Alignments R-Coffee Scaled Consistency Meta-Methods M-Coffee

Open Questions l l Accurately Aligning non transcribed DNA Coping with One Billion Human Genomes

Comparative Bioinformatics l University College Dublin – – – l Berlin Free University – – l Knut Reinert Tobias Rausch Swiss Intitute of Bioinformatics – – l Des Higgins Orla O’Sullivan Iain Wallace (UCD, IE) Ioannis Xenarios Sebastien Morreti Comparative Bioinformatics – – – – Merixell Oliva Giovanni Bussoti Carsten Kemena Emanuele Rainieri Ionas Erb Jia Ming Chang Matthias Zytneki www. tcoffee. org cedric. notredame@crg. es

www. tcoffee. org cedric. notredame@europe. com


Why So Much Interest For Multiple Alignments ? Extrapolation Structure Prediction Motifs/Patterns SNP Analysis Profiles Regulatory Elements Phylogeny Reactivity Analysis

Phylogeny Vs Function: Applications l l Comparative Genomics => New Medium => New Clinical Test

Detecting nc. RNAs in silico: a long way to go… RNAse P

Obtaining the Structure of a nc. RNA is difficult l Hard to Align The Sequences Without the Structure l Hard to Predict the Structures Without an Alignment

R-Coffee: Modifying T-Coffee at the Right Place l Incorporation of Secondary Structure information within the Library l Two Extra Components for the T-Coffee Scoring Scheme – – A new Library A new Scoring Scheme


G-INS-i, H-INS-i and F-INS-i use pairwise alignment information when constructing a multiple alignment. The two options ([HF] -INS-i) incorporate local alignment information and do NOT USE FFT.

Molecular Biology Within the System Biology Era Protein A Interacts with Regulates Inhibits Protein B

Molecular Biology In the 1000 Genomes Era Protein A Interacts with Regulates Inhibits Protein B Variation Within Species: CNVs of A and SNPs Conservation Across Species

System Biology vs Comparative Genomics Systems can Evolve through Selection Systems Biology Systems can be Understood

Phylogeny Vs Function: Applications – Important Application – Possible Many New Genomes – Challenging Too Many New Genomes

3 D-Coffee: Combining Sequences and Structures Within Multiple Sequence Alignments

Comparing Methods MAFFT

Some Benchmark: BB 11 Bali. Base BB 11: 38 highly divergent (less than 25% id) datasets from Bali. Base BB 11: predicts 78% of the results measured on other datasets Blackshield, Higgins

Ph. D Fellowships www. crg. es

What ‘s in a Multiple Sequence Alignment Selection Important Features Are Preserved Evolution Inertia Common Ancestry Shows up In the sequences Phylogenetic Footprint, Evolutionary Trace … Functional Constraint Same Function Same Sequence Convergence

Which Tool for Which Sequence ?

Is it Possible to Compare all Types of Sequences l Non Transcribed World – Genes/Full Genomes l – Promoter Regions l l – Meta-Aligner Motifs Finders Nucleosome l l Lagan, TBA ? ? ? Multiple Genome Aligners – – – Not Very Accurate Very Fast Deal with rearrangements

Is it Possible to Compare all Types of Sequences l RNA Comparison – – l Less Accurate than Proteins Secondary Structures nc. RNA World – – Consan R-Coffee

Is it Possible to Compare all Types of Sequences l Protein Comparisons – – l Very Accurate 3 D-Structure Improves it Protein Aligners – – – Clustal. W T-Coffee 3 D-Coffee

Why So Much Interest For Multiple Alignments ? Extrapolation Structure Prediction Motifs/Patterns SNP Analysis Profiles Regulatory Elements Phylogeny Reactivity Analysis

What’s in a Multiple Alignment ? l The MSA contains what you put inside: – – – l Structural Similarity Evolutive Similarity Sequence Similarity You can view your MSA as: – – – A record of evolution A summary of a protein family A collection of experiments made for you by Nature…

Producing The Right Alignment l Multiple Sequence Alignments Influence Phylogenetic Trees l Choice of Method is not Neutral – – – l Different Methods Different Alignments Different Trees Using The Right Models insures Producing the right Tree

Model Based Alignments vs Naïve Alignments l Naïve Alignment – – – l Model Based Alignments – – l Lexicographic Alignment Maximizing the number of identities At best using a substitution matrix Using a model Protein structure information RNA Structure information Combining/Confronting Modeling methods Template based Alignments – Model based Alignments through the use of Templates

T-Coffee and Model Based Alignments l T-Coffee Algorithm l Expresso: Aligning Protein Structures l R-Coffee: Aligning RNA structures l M-Coffee: Combining methods

T-Coffee and Concistency…

When Sequences Are not Enough 3 D-Coffee and Expresso

3 D-Coffee: Combining Sequences and Structures Within Multiple Sequence Alignments

3 D-Coffee: Combining Sequences and Structures Within Multiple Sequence Alignments

Where to Trust Your Alignments Most Methods Disagree Most Methods Agree

Conclusion l Model Based Alignments Give the best Accuracy l Template based alignment is a very efficient way to turn Naïve aligners into model based aligners l Sequence Alignments are not necessarily reliable over their entire lengths

Manguel M, Samaniego F. J. , Abraham Wald’s Work on Aircraft Suvivability, J. American Statistical Association. 79, 259 -270, (1984)

Building and Using Models 35. 67 Angstrom

Computing the Correct Alignment is a Complicated Problem

Stochastic Optimization

Stochastic Optimization l Exploration of Complex Optimization Problems With Multiple Constraints – – l Generation of Population of Suboptimal Solutions – l Genomic Alignments RNA Alignments Quality=f( optimality ) Specification of Concistency Objective Function of TCoffee

Three Types of Algorithms l Progressive: Clustal. W l Iterative: Muscle l Concistency Based: T-Coffee and Probcons
 l")
T-Coffee and Concistency… l Each Library Line is a Soft Constraint (a wish) l You can’t satisfy them all l You must satisfy as many as possible (The easy ones)
 – l Martin Vingron (1991) – –")
Concistency Based Algorithms: T-Coffee l Gotoh (1990) – l Martin Vingron (1991) – – l Concistency Agglomerative Assembly T-Coffee (2000, Notredame) – – l Dot Matrices Multiplications Accurate but too stringeant Dialign (1996, Morgenstern) – – l Iterative strategy using consistency Concistency Progressive algorithm Prob. Cons (2004, Do) – T-Coffee with a Bayesian Treatment

How Good Is My Method ?

Structures Vs Sequences

Validation Using Bali. Base T-Coffee Results

Too Many Methods for ONE Alignment M-Coffee


Estimating the Accuracy of your MSA

What To Do Without Structures

3 D-Coffee: Combining Sequences and Structures Within Multiple Sequence Alignments

Expresso: Finding the Right Structure Why Not Using Structure Based Alignments

Template Based Multiple Sequence Alignments

Template Based Multiple Sequence Alignments Sources -Structure Templates -Profile -… Template Aligner -Structure -Profile Templates -… Template Alignment Source Template Alignment Remove Templates Library

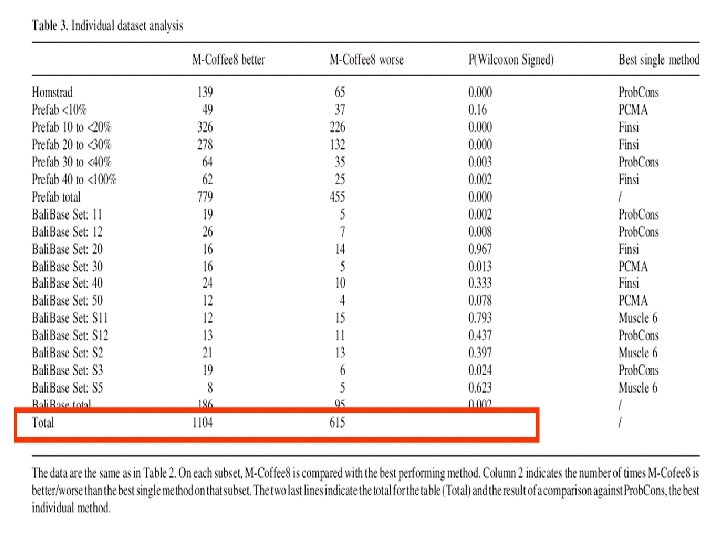
Method Score Templates Prefab Homstrad -------------------------------Clustal. W Matrix ---61. 80 ---Kalign Matrix ---63. 00 ---MUSCLE Matrix ---68. 00 45. 0 -------------------------------T-Coffee Consistency ---69. 97 44. 0 Prob. Consistency ---70. 54 ---Mafft Consistency ---72. 20 ---M-Coffee Consistency ---72. 91 ---MUMMALS Consistency ---73. 10 --------------------------------Clustal-db Matrix Profiles ------PRALINE Matrix Profiles ---50. 2 PROMALS Consistency Profiles 79. 00 ---SPEM Matrix Profiles 77. 00 --------------------------------EXPRESSO Consistency Structures ---71. 9 * T-Lara Consistency Structures ----------------------------------Table 1. Summary of all the methods described in the review. Validation figures were compiled from several sources, and selected for the compatibility. Prefab refers to some validation made on Prefab Version 3. The HOMSTRAD validation was made on datasets having less than 30% identity. The source of each figure is indicated by a reference. *The EXPRESSO figure comes from a slightly more demanding subset of HOMSTRAD (HOM 39) made of sequences less than 25% identical.

Improving The Evaluation

How Do We Perform In The Twilight Zone? l l l Concistency Based Methods Have an Edge Hard to tell Methods Apart Sequence Alignment is NOT solved

More Than Structure based Alignments l Structural Correctness Is Only the Easy Side of the Coin. l In practice MSA are intermediate models used to generate other models: Data Model Type Benchmark Homology Profile Yes Evolution Trees No Structure 3 D-Structure CASP Function Annotation No

Conclusion l Template based Multiple Sequence Alignments l l l Need for new evaluation procedures l l l Projecting any relevant information onto the sequences Using this Information Functional Analysis Phylogenetic Analysis Homology Search (Profiles) Homology Modelling Integrating data Making sure your bits of data can fight with one another

Turning Data into Models Data Columbus, considered that the landmass occupied 225°, leaving only 135° of water (Marinus of Tyre, 70 AD). Columbus believed that 1° represented only 56 miles (Alfraganus, XIth century) He knew there was an island named Japan off the cost of China… Model Circumference of the Earth as 25, 255 km at most, Canary Island to Japan : 3, 700 km (Reality: 12, 000 km. )

The More Structures The Merrier Average Improvement over T-Coffee Struc/Seq Ratio

The Right Mixt of Methods

3 D-Coffee: Combining Sequences and Structures Within Multiple Sequence Alignments

Applications

Looking-Up The DNA Behind The Sequences: PROTOGENE

SAR Analysis l Correlate Alignment Variations with Reactivity Application to the Human Kinome Collaboration with Sanofi-Aventis l Main Issue: l l – Training problem Proper Benchmarking

nc. RNA Multiple Alignments with R-Coffee Laundering the Genome Dark Matter Cédric Notredame Comparative Bioinformatics Group Bioinformatics and Genomics Program

No Plane Today…

nc. RNAs Comparison l And ENCODE said… “nearly the entire genome may be represented in primary transcripts that extensively overlap and include many non-protein-coding regions” l Who Are They? – – l t. RNA, r. RNA, sno. RNAs, micro. RNAs, si. RNAs pi. RNAs long nc. RNAs (Xist, Evf, Air, CTN, PINK…) How Many of them – – – . Open question 30. 000 is a common guess Harder to detect than proteins

nc. RNAs can have different sequences and Similar Structures

nc. RNAs are Difficult to Align l Same Structure Low Sequence Identity l Small Alphabet, Short Sequences Alignments often Non. Significant

Obtaining the Structure of a nc. RNA is difficult l Hard to Align The Sequences Without the Structure l Hard to Predict the Structures Without an Alignment

The Holy Grail of RNA Comparison: Sankoff’ Algorithm

The Holy Grail of RNA Comparison Sankoff’ Algorithm l Simultaneous Folding and Alignment – – l In Practice, for Two Sequences: – – l Time Complexity: O(L 2 n) Space Complexity: O(L 3 n) 50 nucleotides: 100 nucleotides 200 nucleotides 400 nucleotides 1 min. 16 min. 4 hours 3 days Forget about – – Multiple sequence alignments Database searches 6 M. 256 M. 4 G. 3 T.

The next best Thing: Consan l Consan = Sankoff + a few constraints l Use of Stochastic Context Free Grammars – – Tree-shaped HMMs Made sparse with constraints l The constraints are derived from the most confident positions of the alignment l Equivalent of Banded DP

Going Multiple…. Structural Aligners

Game Rules l Using Structural Predictions – – l Produces better alignments Is Computationally expensive Use as much structural information as possible while doing as little computation as possible…

Adapting T-Coffee To RNA Alignments

T-Coffee and Concistency…

T-Coffee and Concistency…

T-Coffee and Concistency…

T-Coffee and Concistency…

Consistency: Conflicts and Information W X Y X Z Y Z Y W Y is unhappy W Z X is unhappy X X X Y Y Y Z W Z Fully Consistent More Reliable W Partly Consistent Less Reliable Z

RNA Sequences Consan or Mafft / Muscle / Prob. Cons RNAplfold Primary Library Secondary Structures R-Coffee Extension R-Coffee Extended Primary Library R-Score Progressive Alignment Using The R-Score
=MAX(TC-Score(CC), TC-Score (GG)) C C G G")
R-Coffee Scoring Scheme R-Score (CC)=MAX(TC-Score(CC), TC-Score (GG)) C C G G

Validating R-Coffee

RNA Alignments are harder to validate than Protein Alignments l Protein Alignments Use of Structure based Reference Alignments l RNA Alignments No Real structure based reference alignments – – The structures are mostly predicted from sequences Circularity

Brali. Base and the Brali. Score l Database of Reference Alignments l 388 multiple sequence alignments. l Evenly distributed between 35 and 95 percent average sequence identity l Contain 5 sequences selected from the RNA family database Rfam l The reference alignment is based on a SCFG model based on the full Rfam seed dataset (~100 sequences).

Brali. Base SPS Score RFam MSA SPS= Number of Identically Aligned Pairs Number of Aligned Pairs
))…((.")
Brali. Base: SCI Score Covariance R N A p f o l d (((…)))…((. . )) DG Seq 1 (((…)))…((. . )) DG Seq 2 (((…)))…((. . )) DG Seq 3 (((…)))…((. . )) DG Seq 4 (((…)))…((. . )) DG Seq 5 (((…)))…((. . )) DG Seq 6 RNAlifold SCI= (((…)))…((. . )) ALN DG Average DG Seq X Cov DG ALN

BRali. Score Braliscore= SCI*SPS

RM-Coffee + Regular Aligners Method Avg Braliscore Net Improv. direct +T +R -----------------------------Poa 0. 62 0. 65 0. 70 48 154 Pcma 0. 62 0. 64 0. 67 34 120 Prrn 0. 64 0. 61 0. 66 -63 45 Clustal. W 0. 65 0. 69 -7 83 Mafft_fftnts 0. 68 0. 72 17 68 Prob. Cons. RNA 0. 69 0. 67 0. 71 -49 39 Muscle 0. 69 0. 73 -17 42 Mafft_ginsi 0. 70 0. 68 0. 72 -49 39 -----------------------------RM-Coffee 4 0. 71 / 0. 74 / 84

How Best is the Best…. Method vs. R-Coffee-Consan vs. RM-Coffee 4 Poa 241 *** 217 *** T-Coffee 241 *** 199 *** Prrn 232 *** 198 *** Pcma 218 *** 151 *** Proalign 216 *** 150 ** Mafft fftns 206 *** 148 * Clustal. W 203 *** 136 *** Probcons 192 *** 128 * Mafft ginsi 170 *** 115 Muscle 169 *** 111 M-Locarna 234 *** 183 ** Stral 169 *** 62 Foldalign. M 146 61 Murlet 130 * -12 Rnasampler 129 * -27 T-Lara 125 * -30

Range of Performances Effect of Compensated Mutations

Conclusion/Future Directions l T-Coffee/Consan is currently the best MSA protocol for nc. RNAs l Testing how important is the accuracy of the secondary structure prediction l Going deeper into Sankoff’s territory: predicting and aligning simultaneously

Credits and Web Servers l Andreas Wilm Des Higgins Sebastien Moretti Ioannis Xenarios Cedric Notredame l CGR, SIB, UCD l l www. tcoffee. org cedric. notredame@europe. com

Prank Vs Prank

Prank Vs Prank Gop=0 Gep=0

Prank Vs Prank The reconstruction of evolutionary homology -- including the correct placement of insertion and deletion events -- is only feasible for rather closely-related sequences. PRANK is not meant for the alignment of very d i v e r g e d p r o t e i n s e q u e n c e s. I f sequences are very different, the correct homology cannot be reconstructed with confidence and http: //www. ebi. ac. uk/goldmansrv/prank/

Do Benchmarks All Tell the same story? Based on
 ∑k P(xi ~")
Probcons: Different Primary Library Score(xi ~ yj | x, y, z) ∑k P(xi ~ zk | x, z) P(zk ~ yj | z, y) Score=S (MIN(xz, zk))/MAX(xz, zk)
- Slides: 162