Caso Clnico Internato em Pediatria Hospital Regional da





, eupnéico, afebril, acianótico, anictérico, sorridente, ativo e")

")




: 12’’; 87, 1% Ø EAS (01/10/06): dens → 1005;")

















. Ø Ácido láctico é o substrato")

























 Microscopia: arquitetura lobular preservada e espaços portas c/ seus constituintes permeado")


. Ø Alto teor")




- Slides: 66
Caso Clínico Internato em Pediatria – Hospital Regional da Asa Sul (Escola Superior de Ciências da Saúde) INTERNO: Vinicius Silveira Amaral ORIENTADORA: Elisa de Carvalho
ANAMNESE Ø História colhida em 02/10/06 Ø H. S. N. , masculino, 2 anos e 3 dias, procedente de Águas Lindas - GO
ANAMNESE Ø QP: Vômitos há ± 48 horas. Ø HDA: Paciente com diagnóstico prévio de glicogenose, em acompanhamento com a gastropediatria, vem encaminhado do HRAN com o seguinte relatório.
ANAMNESE Ø HDA: Relatório do HRAN: iniciou quadro de vômitos e apatia há 48 horas. Mãe procurou pelo HRAN, onde foi realizada glicemia capilar (45 mg%). Em casa, foi atendido pelo SAMU, que constatou glicemia de 18 mg%, sendo feito soro glicosado (SIC). No momento: paciente apresenta-se sem queixas. Diurese e evacuações preservadas e sem anormalidades. Ótima aceitação da dieta. Nega febre, diarréia ou vômitos. Não faz uso de medicações regularmente.
ANAMNESE Antecedentes Ø Glicogenose diagnosticada em 02/09/05, aos 1 a 1 m de vida (em acompanhamento com a gastropediatria desde 31/10/05, quando foi encaminhado pelo HRT, após vários episódios de hipoglicemia de difícil controle). Ø Não estava realizando as consultas regularmente no ambulatório de gastropediatria.
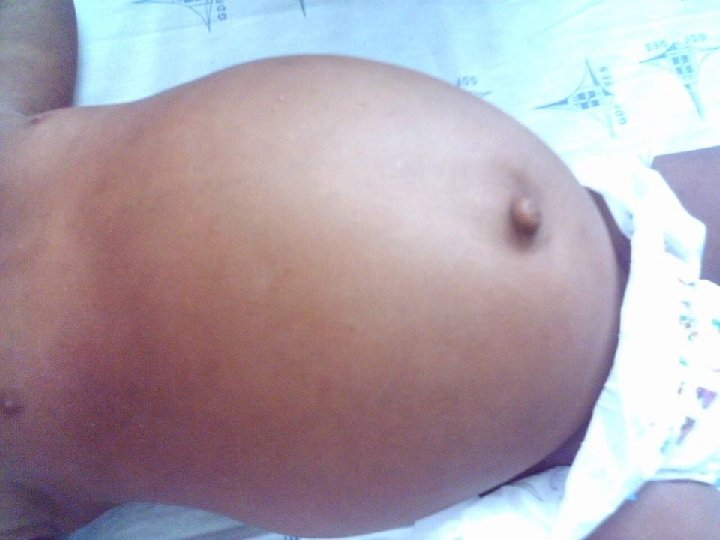
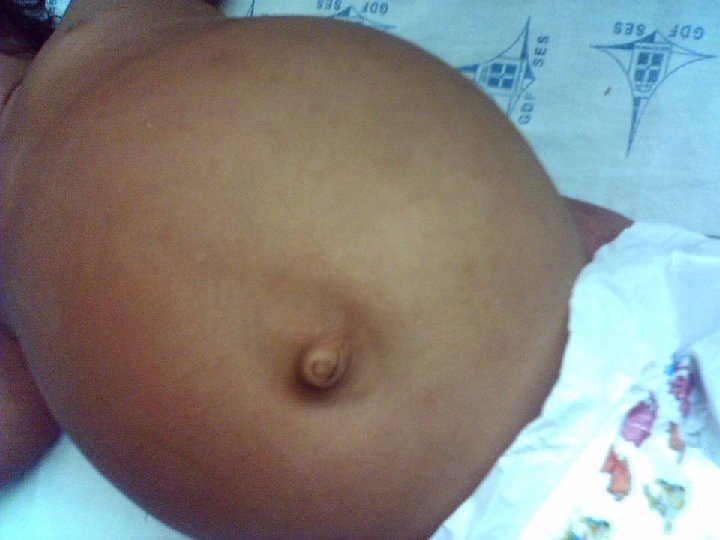
EXAME FÍSICO Ø BEG, hidratado, hipocorado (+/+4), eupnéico, afebril, acianótico, anictérico, sorridente, ativo e reativo. Peso: 9 kg Ø ACV: RCR em 2 T, BNF, s/ sopro. FC: 100 bpm. Ø AR: MV+ bilateralmente, s/ RA, s/ esforço respiratório. FR: 26 irpm.
EXAME FÍSICO Ø ABD: semi-globoso, depressível, RHA+, timpânico, indolor, fígado a ± 3, 5 cm do RCD, baço não palpável. Ø Ext: MMII ressecados. Pulsos palpáveis, simétricos, boa amplitude. Bem perfundidas. Discreta lesão crostosa em região tibial inferior D, s/ sinais flogísticos.
HIPÓTESE DIAGNÓSTICA Ø GLICOGENOSE (TIPO ? ? )
EXAMES COMPLEMENTARES Hb Ht Leuco Seg Bast Linf Mon Eos Plaq 27/10/05 01/10 02/10 03/10 10, 6 12, 1 13, 2 34, 7 35, 7 39, 7 5400 15400 9400 11900 50 69 55 45 08 01 00 00 35 27 36 50 06 02 03 01 01 01 06 03 470000 454000 609000 472000
EXAMES COMPLEMENTARES Glic Creat Uréia Ca 2+ Na+ K+ TGO TGP Trig 27/10/05 01/10 02/10 03/10 11 84 55 87 0, 3 0, 4 16 15 14 8, 6 9, 7 10 133 132 135 4, 4 4, 2 49 27 32 20 14 23 126 252 106 158
EXAMES COMPLEMENTARES BT BD BI Cl. FA GGT Ác. úr PT ALB 27/10/05 01/10 02/10 03/10 0, 17 0, 2 0, 04 0, 13 102 100 104 1525 488 33 19 3, 72 3, 3 5, 8 7, 6 3, 8 4, 7
GASOMETRIAS p. H p. CO 2 p. O 2 ABE Lactato HCO 3 Hb Glicose 27/10/05 03/10/06 7, 32 7, 4 45 26 97 71, 2 -2 -7, 3 34 22 18, 9 10, 6 105
OUTROS EXAMES Ø TAP (26/10/05): 12’’; 87, 1% Ø EAS (01/10/06): dens → 1005; p. H → 7; CED → 2 -3; LEUCO → 2 p/c Ø Rx de tórax (02/10/06): normal Ø PCR (03/10/06): 0, 17
GLICEMIAS CAPILARES Ø 02/10/06: 38 mg% Ø 03/10/06: 27 mg%, 22 mg% e 33 mg% Ø 04/10/06: 19 mg%
Hipoglicemia + Hepatomegalia
METABOLISMO DO GLICOGÊNIO Ø Glicose →hepatócito→G-6 -P→ 5 vias. Ø Vias: glicólise (piruvato e lactato); ciclo das pentoses (NADPH 2 e CO 2); formação de gluconato; glicose livre; síntese de glicogênio. Ø Glicogênio: presente no fígado e Mm. Composto por glicose. Frutose, galactose e AA são potencialmente glicogênicos. Formação é favorecida pela insulina.
METABOLISMO DO GLICOGÊNIO
METABOLISMO DO GLICOGÊNIO
METABOLISMO DO GLICOGÊNIO Ø Deposição de glicogênio: inibição da fosforilase e estimulação da glicogênio sintetase. Ø Fígado libera glicose em situações de estresse ou quando níveis séricos de glicose estão reduzidos.
GALACTOSEMIA Ø Deficiência da Galactose-1 -fosfato-UDPtransferase. Ø Autossômica recessiva. Ø Incidência de 1: 50000 nascidos vivos. Ø Efeitos tóxicos, agudos e crônicos, no fígado e em outros órgãos. Ø Início após introdução de dieta láctea.
GALACTOSEMIA Ø Q. C. : hepatomegalia, icterícia, vômitos, hipodesenvolvimento, distensão abdominal, convulsões, hipoglicemia e catarata. Ø Laboratório: ↑ da galactose sérica e urinária, bilirrubinas e aminotransferases; presença de substâncias redutoras na urina; hipoalbuminemia; coagulopatia; acidose hiperclorêmica.
GALACTOSEMIA Ø Aumento de óbitos nos 1 os. meses de vida, por sepse por E. coli. Ø Se tratado precocemente, a cirrose é um evento raro.
DEFICIÊNCIA DA FRUTOSE-1, 6 DIFOSFATASE Ø Hipoglicemia por jejum ou ingestão de frutose. Ø Sintomas graves nos 1 os. dias de vida (acidose metabólica grave). Ø Irritabilidade, sonolência, apnéia, taquicardia, hipotonia e hepatomegalia. Ø Descompensação: doença aguda, jejum ou aumento da ingestão de frutose (hipoglicemia grave e convulsões).
INTOLER NCIA HEREDITÁRIA À FRUTOSE Ø Deficiência de Frutose-1 -fosfato Aldolase. Ø Autossômica recessiva. Ø Incidência de 1: 30000 nascidos vivos. Ø Ingestão de frutose leva a alterações no fígado, rins e intestino. Ø Acúmulo de frutose-1 -fosfato (inibe gliconeogênese e glicogenólise).
INTOLER NCIA HEREDITÁRIA À FRUTOSE Ø Início: introdução de sacarose/frutose na dieta. Ø Irritabilidade, vômitos, letargia, convulsões, coma, sudorese, náuseas, icterícia, edema, diarréia, hipodesenvolvimento, sepse, coagulopatia, hepatomegalia. Ø Aversão a alimentos doces. Ø Se não houver vômitos, há grande probabilidade de que não seja a doença.
INTOLER NCIA HEREDITÁRIA À FRUTOSE Ø Laboratório: hipoglicemia; hipofosfatemia; ↑ das aminotransferases, bilirrubinas, da excreção dos uratos; hiperuricemia; acidose hiperclorêmica; hiperlactatemia; hipocalemia; hipermagnesemia; anemia e trombocitopenia. Ø Diagnóstico: confirmação pela medida direta da frutose-1 -Paldolase no tecido hepático. Ø Esteatose persistente: ingestão de fontes ocultas de frutose. Ø Cirrose se não houver dieta c/ exclusão de frutose.
GLICOGENOSE Ø Erro inato do metabolismo (alteração da concentração ou estrutura do glicogênio no organismo). Ø Classificada em 12 tipos. Ø Classificação é baseada nos defeitos enzimáticos específicos e nos tecidos comprometidos.
GLICOGENOSE Ø Tipos I, III, IV, VI, IX e XI têm comprometimento hepático. Ø Hipoglicemia de jejum + acidose + hepatomegalia + retardo do crescimento. Ø Características histopatológicas são muito semelhantes. Impossível correlação morfológica com o tipo de deficiência enzimática (exceto no tipo IV).
GLICOGENOSE TIPO I Ø Deficiência de G-6 -Pase. Ø 25% das glicogenoses. Ø Autossômica recessiva. Ø 5 subtipos (Ia, Ia. SP, Ib, Ic, Id). Ø Incidência de 1: 100000 nascidos vivos.
GLICOGENOSE TIPO I Ø Não há liberação de glicose pelo fígado. Ø Metabolismo da frutose e galactose não fornecerá glicose livre. Ø Hipoglicemia de jejum é freqüente (cetose não é proeminente).
GLICOGENOSE TIPO I Ø Hipoglicemia→insulina↓ e glucagon↑ (cronicidade). Ø Ácido láctico é o substrato para o metabolismo cerebral (ação protetora para o SNC). Ø Hiperuricemia: ↓ depuração renal do urato e ↑ produção do ác. úrico (gota, nefrolitíase e nefropatia).
GLICOGENOSE TIPO I Ø Hiperlipidemia: ↑ produtos glicolíticos → ↑ triglicerídeos e colesterol. Ø Hepatomegalia: esteatose hepática e acúmulo de glicogênio nos hepatócitos. Ø Não há maior risco de doença isquêmica precoce.
GLICOGENOSE TIPO I Glicogenose Ia Ø Hipoglicemia de jejum + hepatomegalia. Ø Observada no período neonatal. Ø Palidez, fome excessiva, sudorese, convulsões, obesidade troncular, face de boneca, retardo estatural, musculatura pouco desenvolvida. Ø Distúrbios da agregação plaquetária.
GLICOGENOSE TIPO I Glicogenose Ia Ø Xantomas, osteoporose, fraqueza, cefaléia, taquipnéia e mal-estar. Ø Nefromegalia (nefropatia progressiva, proteinúria, HAS). Ø Não evolui com cirrose ou insuficiência hepática. Ø Desenvolvimento de adenomas (hemorragia e transformação maligna).
GLICOGENOSE TIPO I Glicogenose Ib Ø Semelhante ao tipo Ia. Ø ↓ e disfunção de neutrófilos (infecções piogênicas, gengivoestomatite, DII e periodontite). Ø Medula óssea: hiperplasia da série mielóide e atraso na maturação de precursores dos neutrófilos.
GLICOGENOSE TIPO I Diagnóstico Laboratorial Ø Hipoglicemia de jejum; ↑ de ác. láctico, ác. úrico, colesterol, ác. graxos, triglicerídeos, fosfolipídios e aminotransferases. Ø Teste de tolerância ao glucagon: tipo I → s/ resposta ao glucagon em jejum ou após alimentação; tipo III → há resposta+, se realizado após a dieta.
GLICOGENOSE TIPO I Diagnóstico Laboratorial Ø Tipo Ia: atividade da G-6 -Pase está ausente no tecido hepático fresco e congelado. Ø Tipo Ib: atividade da G-6 -Pase é normal no tecido congelado.
GLICOGENOSE TIPO I Histologia Ø Células hepáticas tumefeitas; hepatócitos pálidos e c/ grandes vacúolos de gordura no citoplasma; membrana citoplasmática está espessada; parênquima c/ aspecto de mosaico; fenômeno de hiperglicogenose nuclear; pode haver fibrose (mais comum nos tipos III, IV, VI, IX e XI).
GLICOGENOSE TIPO III Ø Autossômica recessiva. Ø Deficiência da enzima desramificadora. Ø Incidência de 1: 50000 a 1: 100000 nascidos vivos. Ø Tipo IIIa: perda generalizada da atividade enzimática (80% dos casos; miopatia nos adultos). Ø Tipo IIIb: atividade enzimática preservada nos Mm. (s/ miopatia nos adultos).
GLICOGENOSE TIPO III Quadro Clínico Ø Manifestações menos intensas que no tipo I. Ø Hepatomegalia + déficit de crescimento. Ø Menos hipoglicemia de jejum; comprometimento muscular é maior. Ø Miopatia (↑ da CPK sérica).
GLICOGENOSE TIPO III Quadro Clínico Ø Cardiomiopatia após os 30 anos. Ø Pode haver glicose liberada pela fosforilase (glicemia mantida por gliconeogênese de AA e metabolismo da galactose e frutose). Ø Adulto: assintomático. Ø Há cetose e cetonúria de jejum; não há acidose láctica importante.
GLICOGENOSE TIPO III Quadro Clínico Ø ↑ de colesterol, triglicerídeos e aminotransferases. Concentração de urato é normal. Pode haver fibrose hepática. Ø Puberdade atrasada (altura final é preservada). Ø Envolvimento hepático é autolimitado.
GLICOGENOSE TIPO III Complicações Ø Perda e atrofia muscular; hipertensão porta secundária à cirrose hepática. Ø Hemorragia digestiva e carcinoma hepatocelular. Ø Prevalência de adenoma é de 25%
GLICOGENOSE TIPO III Diagnóstico Ø Determinação da atividade da enzima desramificadora no fígado, Mm. , eritrócitos, leucócitos e fibroblastos. Ø Identificação de mutações. Ø Histopatologia: semelhante ao tipo I, c/ hiperglicogenose nuclear periportal, menor fibrose periportal e esteatose. Acúmulo de glicogênio no fígado e Mm. (tipo IIIa); acúmulo de glicogênio no fígado (IIIb).
GLICOGENOSE TIPO IV Ø Autossômica recessiva. Ø Forma menos comum. Ø Deficiência da enzima ramificadora (acúmulo de amilopectina). Ø Estrutura do glicogênio está alterada.
GLICOGENOSE TIPO IV Quadro Clínico Ø Atraso do crescimento, distensão abdominal e hepatoesplenomegalia nos 1 os. meses de vida. Ø Há casos congênitos (hidropsia fetal). Ø Cirrose e hipertensão porta no final do 1º. ano → insuficiência hepática e morte antes dos 4 anos de vida.
GLICOGENOSE TIPO IV Quadro Clínico Ø Cardiomiopatia e carcinoma hepatocelular podem complicar o quadro. Ø Hipotonia e atrofia muscular. Ø É rara a hipoglicemia (metabolismo do carboidrato é normal). Ø ↑ de aminotransferases e bilirrubina direta.
GLICOGENOSE TIPO IV Diagnóstico Ø Deficiência da atividade enzimática no fígado, leucócitos, Mm. , eritrócitos ou fibroblastos. Ø Histopatologia: acúmulo de substância anormal (PAS+ diastase -resistente) no fígado e Mm. Acúmulo de material pálido, hialino ou vacuolado por todo o lóbulo hepático (mais intenso na periferia).
GLICOGENOSE TIPO IV Diagnóstico Ø Material PAS+ diastase-resistente é semelhante aos depósitos da deficiência de alfa-1 -antitripsina. Ø Estágio avançado: acúmulos nodulares birrefringentes de material diferente, hialino e fibrilar, difusamente distribuído nos lóbulos hepáticos.
GLICOGENOSE TIPO VI Ø Rara. Ø Deficiência da fosforilase no fígado. Ø Possivelmente seja herança autossômica recessiva. Ø Evolução benigna; características semelhantes ao tipo III.
GLICOGENOSE TIPO VI Ø Hepatomegalia; atraso de crescimento; tolerância ao jejum; ↑ dos níveis séricos de ác. úrico, colesterol, triglicerídeos, aminotransferases. Ø Hipoglicemia e cetose são discretas. Ø Histopatologia: mosaico de hepatócitos distendidos. Dilatação celular heterogênea (principalmente na periferia do lóbulo). Membranas grosseiras e onduladas. Vacúolos citoplasmáticos e discretos septos nas áreas portais, mas s/ hiperglicogenose.
GLICOGENOSE TIPO VI Ø Microscopia eletrônica: partículas citoplasmáticas de glicogênio que deslocam as organelas celulares. Ø Tecido muscular é normal. Ø Há relatos de carcinoma hepatocelular, mas s/ adenoma.
GLICOGENOSE TIPO IX Ø Deficiência da fosforilase quinase. Ø Tipo IXa: mais freqüente; acomete o fígado; herança ligada ao sexo. Ø Tipo IXb: semelhante ao tipo IXa; herança autossômica recessiva. Ø Tipo IXc: comprometimento hepático e muscular; herança autossômica recessiva.
GLICOGENOSE TIPO IX Ø Q. C. : hepatomegalia; atraso de crescimento; ↑ de triglicerídeos, colesterol e aminotransferases; hipoglicemia; face de boneca; fraqueza muscular no tipo IXc. Assintomáticos quando adultos. Fibrose hepática, cirrose e carcinoma hepatocelular são raros. Ø Diagnóstico: atividade da fosforilase quinase nos eritrócitos e leucócitos. Ø Histopatologia: hepatócitos distendidos por glicogênio. Septos proeminentes, inflamação mínima. S/ hiperglicogenose nuclear.
GLICOGENOSE TIPO XI Ø Rara; autossômica recessiva. Ø Defeitos no GLUT-2. Ø Disfunção tubular renal proximal, redução da utilização de glicose e galactose, acúmulo de glicogênio no fígado e rins. Ø Q. C. : atraso no crescimento, raquitismo, hepatomegalia e nefromegalia.
GLICOGENOSE TIPO XI Ø Laboratório: glicosúria, aminoacidúria, perda de bicarbonato, hipofosfatemia, ↑ de FA sérica. Hipoglicemia e hiperlipidemia. Aminotransferases, lactato e ác. úrico plasmáticos estão normais. Ø Intolerância no TTOG (perda funcional de GLUT 2 → impede a captação hepática). Ø Biópsia hepática: acúmulo acentuado de glicogênio nos hepatócitos e células tubulares renais proximais, devido ao transporte alterado da glicose para fora desses órgãos.
BIÓPSIA HEPÁTICA (02/09/05) Microscopia: arquitetura lobular preservada e espaços portas c/ seus constituintes permeado por leve infiltrado misto. PARÊNQUIMA: esteatose, macro e microgoticular, hepatócitos c/ citoplasma amplo e distendido, c/ material PAS+ que desaparece quando utiliza-se a diastase. Sinusóides focalmente inaparentes. Diagnóstico histopatológico: Hepatopatia metabólica. OBS. : alterações compatíveis c/ glicogenose.
CONCLUSÃO GLICOGENOSE TIPO I ou III Hipoglicemia pouco acentuada, colesterol, TGO e ácido úrico normais Glicogenose Tipo III Ausência de queixas musculares Glicogenose Tipo IIIb
TRATAMENTO Ø Dieta faz parte do tratamento. Ø Pacientes c/ hipoglicemia devem ter nutrição enteral noturna. Ø Cereais crus têm mais eficácia nas crianças maiores. Ø Amido de milho cru: 2 g/Kg, 4 x/dia. Cças menores de 2 anos, podem não apresentar digestão adequada. Recomenda-se a introdução após 8º. mês de vida.
TRATAMENTO Ø S/ restrição de frutose e galactose (gliconeogênese está normal). Ø Alto teor de carboidratos melhoram a glicemia, hepatomegalia, fraqueza muscular e falência do crescimento. Ø Administrar proteínas em grande quantidade.
TRATAMENTO Ø Dieta hiperprotéica: 45% de CHO, 30% de lipídios e 25% de proteínas. Ø 6 meses-3 anos: 5 -6 g/Kg/dia de proteína. Ø 4 -10 anos: 4 -5 g/Kg/dia de proteína. Ø Maiores de 11 anos: 3 g/Kg/dia de proteína.
TRATAMENTO Ø Monitorar peso, altura, aminotransferases e triglicerídeos. Ø Miopatia pode ser atenuada com nutrição enteral hiperprotéica.
REFERÊNCIAS BIBLIOGRÁFICAS FERREIRA, Cristina T. ; CARVALHO, Elisa de; SILVA, Luciana R. Glicogenoses. In: FAGUNDES, Eleonora D. T. ; FERREIRA, Alexandre R. ; ROQUETE, Mariza L. V. Gastroenterologia e hepatologia em pediatria: diagnóstico e tratamento. Rio de Janeiro: MEDSI EDITORA MÉDICA E CIENTÍFICA LTDA, 2003. cap. 49, p. 645 -657. FERREIRA, Cristina T. ; CARVALHO, Elisa de; SILVA, Luciana R. Outras doenças metabólicas do fígado. In: BEZERRA, Jorge A. Gastroenterologia e hepatologia em pediatria: diagnóstico e tratamento. Rio de Janeiro: MEDSI EDITORA MÉDICA E CIENTÍFICA LTDA, 2003. cap. 50, p. 660 -663. SCRIVER, Charles R. et al. Glycogen Storage Diseases In: CHEN, Yuan-Tsong; BURCHELL, Ann; The metabolic and molecular bases of inherited disease. New York: THE MCGRAW-HILL COMPANIES, INC, 1995. cap. 24, p. 935 -965. 7 ed. Volume 1. BRAUNWALD, Eugene et al. Doenças de depósito de glicogênio e outros distúrbios hereditários do metabolismo dos carboidratos. In: CHEN, Yuan-Tsong. Harrison: medicina interna. Rio de Janeiro: MCGRAW-HILL INTERAMERICANA DO BRASIL LTDA, 2001. cap. 350, p. 2426 -2433. 15 ed. Volume 2.
OBRIGADO