Case presentation Dr Waceke N Kombe HISTORY M

Case presentation Dr Waceke N Kombe

HISTORY • M. H. N • Age: 1 year 3 months • C/O Abdominal Distension for 1 week. No yellowness of eyes, not easily fatigued, passing urine normally. • No vomiting, No diarrhoea. Feeding has been good. • Seen at another hospital, Ultrasound done and reported to show enlarged liver and spleen.

HISTORY • Mother reports that she lost a sibling at 23 months of age who started to have Abdominal Distension and was diagnosed to have hepatosplenomegaly. An autopsy was done but the results were inconclusive. One surviving sibling, 2 years 8 months, no known medical conditions. • This is the first admission. Born at term. BWT 3. 0 Kg. c/s due to a previous scar. She walks with support.

EXAMINATION • O/E low set ears with flat nasal bridge. No simian crease. Short stature. • Current weight 10 kg Length 75. 5 cm, H/Cir 45 cm at 2 years. • Apex beat 5 th ICS MCL. Normal heart sounds. • Abdomen looked distended uniformly. No scars, no superficial veins. Bowel sounds normal. Hepatosplenomegaly noted. Liver 6 -8 cm below costal margin. Spleen 4 cm below costal margin. Difficult to assess for ascites. Normal bowel sounds. Kidneys not palpable.

Growth chart

INVESTIGATIONS • FBC Hb 9. 8, MCV 57. 1, MCH 18. 9, PLT 134, WBC 6. 98 (L 1. 9, N 3. 93). • HIV- NEGATIVE • HEB B s Ag , CMV Ig M- NEGATIVE • No evidence of Beta Thalassemia or Hemoglobinopathy • LFT’S • GAMMA GT 47 (N=22) • AST 80 (N=48)
 • U/E/C NORMAL • THYROID MARKERS NORMAL • ALPHA")
INVESTIGATIONS • FERRITIN 141 (N=67) • U/E/C NORMAL • THYROID MARKERS NORMAL • ALPHA FETO PROETIN NORMAL • URINE REDUCING SUBSTANCES- NEGATIVE

Abdominal ultrasound • The liver is grossly enlarged having a span of 13. 3 cm X 8. 4 cm X 9. 4 cm. Normal echotexture, intrahepatic bile ducts not dilated. Gall bladder is normal, no gall stones. • Spleen is markedly enlarged measuring 10. 8 x 12. 5 x 9. 82 cm. It has a normal echotexture, no obvious portal varices. • Adrenal, kidney and pancreas are normal. No bowel lesion seen. There is no ascites.

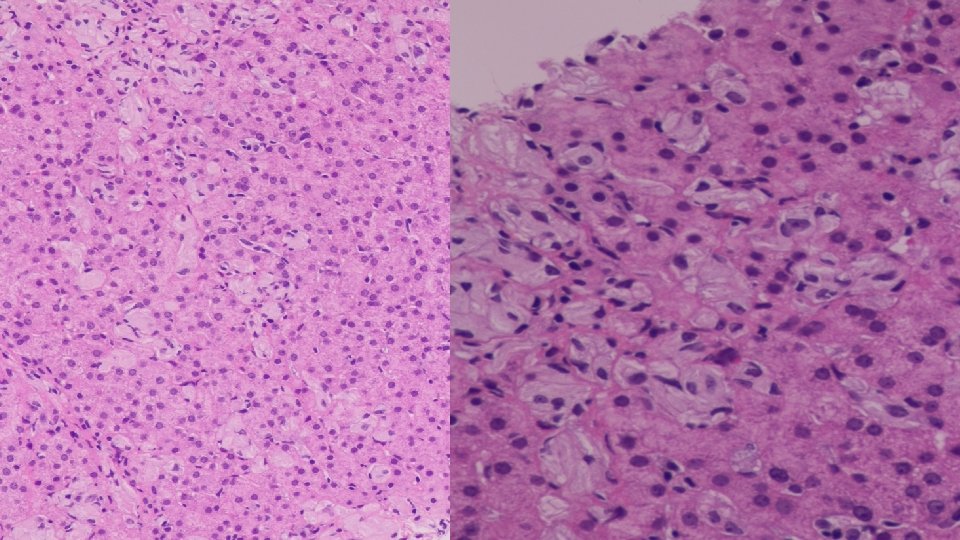
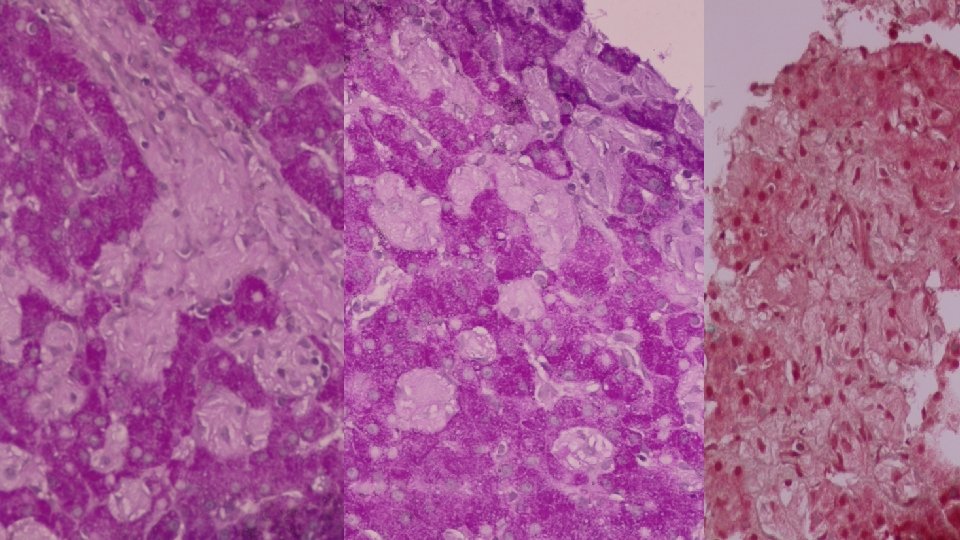
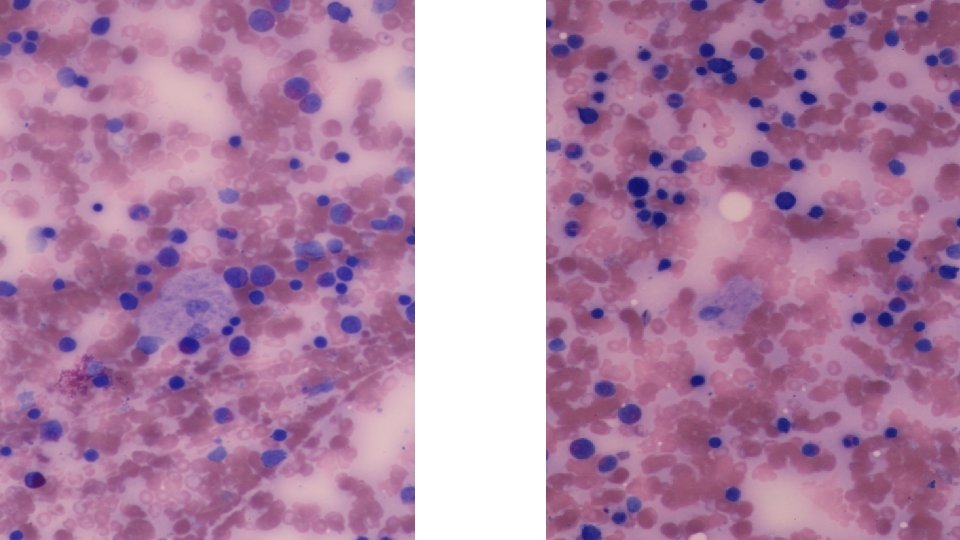
• BONE MARROW ASPIRATE • Patchy infiltration by large cells with abundant wrinkled cytoplasm, some multinucleated. Features of a lysosomal storage disease. • LIVER BIOPSY • Reactive hepatocytes with sinusoidal expansion by enlarged Kupfer cells with abundant wrinkled paper cytoplasm. Features favour Gauchers Disease.

• ENZYME ASSAYS. • Beta Glucosidase activity in serum: not detectable. Deficient activity of beta glucosidase noted in leucocytes.

What could this be?

GAUCHERS DISEASE

• Lysosomal lipid storage disease. • One of the most common lysosomal storage diseases. • Autosomal recessive. • Incidence of Type 1(99% of cases) among Ashkenazi Jews 1 -1000 live births; 1: 50, 000 European Jewish population. • 3 clinical subtypes: • Type 1 – adult, nonneuronopathic form • Type 2 - infantile or acute neuronopathic form • Type 3 - juvenile or subacute neuronopathic form

• Gaucher disease results from the deficient activity of Glucosylceramide β glucosidase. • This results in the accumulation of the substrate glucosylceramide. • Accumulation occurs in the reticuloendothelial system. The progressive deposition results in infiltration of the bone marrow, progressive hepatosplenomegaly, and skeletal complications.

Pathophysiology of Gauchers disease

GENETICS • Deficiency encoded by a gene located on chromosome 1 q 21 -q 31. • Four mutations account for approximately 95% of mutant alleles among Ashkenazi Jews. • • N 370 S L 444 P 84 ins. G IVS 2+2

CLINICAL MANIFESTATIONS TYPE 1 • Age of onset- variable. Early childhood to adolescent. • Easy bruising- thrombocytopenia; chronic fatigue- anaemia; hepatomegaly, splenomegaly and bone pain. • Pulmonary involvement • Growth retardation • Bone pain, pathologic fractures, pseudosteomyelitis, lytic lesions, osteosclerosis, bone crises with severe pain • Radiologic evidence of skeletal involvement- Erlenmeyer flask deformity of the distal femur

Erlenmeyer flask deformity. Modelling abnormality. So named because of its similarity to the glass flask invented by a German scientist.

TYPE 2 • Rare form • Rapid neurodegenerative course with extensive visceral involvement and death within the first years of life. • Presents in infancy with increased tone, strabismus, hepatosplenomegaly • Failure to thrive and stridor • Psychomotor regression. Death typically occurs secondary to respiratory compromise

TYPE 3 • Presents with clinical manifestations that are intermediate to those seen in types 1 and 2, with presentation in childhood and death by age 10 -15 years. • More common among the Swedish population • Neurologic involvement is present.

INVESTIGATIONS • Bone marrow. Will reveal Gaucher cells. • Enzyme assays. Glucocerebrosidase activity in isolated leucocytes or cultured fibroblasts. • Gene mutation identification

WHAT IS A GAUCHER CELL • The Gaucher cell results from the accumulation of excessive glucocerebroside in cells of the monocyte-macrophage system. • Pushes the nucleus to the periphery

TREATMENT • Enzyme replacement therapy. Recombinant human glucocerebrosidase. 60 IU/kg IV infusion every other week. Reverses most symptoms ( organomegaly, hematologic indices, bone pain) • Velaglucerase alfa • Taliglucerase alfa • Enzyme replacement does not alter the neurologic progression of patients with Gaucher disease types 2 and 3. • Oral substrate reduction agents- miglustat • Bone marrow transplantation. Curative but associated with significant morbidity.
- Slides: 26