Cancers of the Bone Marrow A Clinical Perspective

Cancers of the Bone Marrow: A Clinical Perspective Mary Ward, RN, BS, CTR

Objectives • To increase the Cancer Registrar’s knowledge of the disease process of select cancers of the bone marrow from a clinical perspective • Discuss the pathogenesis, clinical evaluation and treatment options of AML, APL, MDS and Multiple Myeloma • Describe clinical distinctions that correspond with some of the ICD-0 -3 codes for patients diagnosed with AML

What does the Bone Marrow Do? • Produces all the elements of the blood – RBCs – Platelets – WBCs • Proliferating marrow is found in certain areas of the body of adults

Cancers that Affect the Bone Marrow • • Myelodysplastic Syndromes Leukemias Lymphomas* Plasma Cell Myelomas

Case Presentation: JF • 68 yo wmm presents to PCP – c/o fatigue, malaise x 6 weeks, ill-defined fevers and intractable infection • infection treated with abx x 2; • weight loss of 10 lbs, decreased appetite, easy bruising on his extremities • PE revealed wm in no acute distress; presents with nonproductive cough, fever of 101. 9 o, no lymphadeopathy, noted several small bruises on LT arm and RT leg; no ictera, no jaundice;

Case Presentation: JF • • Chest x-ray CT abd Labs: CBC with differential and CMP Peripheral blood smear

Case Presentation: JF • JF is admitted to the inpatient oncology unit at Home Base Hospital • Ongoing diagnostic workup would include: – Uric Acid level – Bone Marrow biopsy and aspirate – Leukemia and Lymphoma panel – HLA typing – Type and Cross for transfusions • Consult to Heme/Onc

AML: Defined • A group of hematopoietic neoplasms that affect the precursor cells of the myeloid cell line • The proliferation of the precursor cells causes a reduced capacity of the mature cells to develop

Epidemiology and Risk Factors • Most common acute leukemia in adults • Median age at diagnosis is 65 years old • Male to female ratio of 5: 3

Risk Factors • Environmental – Chemicals – Radiation – Tobacco – chemotherapy drugs • Genetic abnormalities – Trisomy 21, Fanconi’s anemia, Bloom’s syndrome

AML: Diagnostic Workup • • Peripheral blood smear Bone marrow aspiration and biopsy* Cytogenetics* Immunophenotyping*

AML Classification FAB WHO Histology Code M 0 AML with minimal differentiation 9872/3 M 1 AML without maturation 9873/3 M 2 AML with maturation 9874/3 M 3 Acute promeylocytic leukemia with t(15: 17) q(22; q 12) PML-RARA 9866/3 M 4 Acute myelomonocytic leukemia 9867/3 M 5 a Acute monoblastic/acute monocytic leukemia 9891/3 M 5 b Acute monoblastic/acute monocytic leukemia 9891/3 M 6 Acute erythroid leukemia M 7 Acute megakaryoblastic leukemia 9910/3
(q 22; 122), Favorable prognosis in")
AML with Recurrent Genetic Abnormalities AML with t(8; 21)(q 22; 122), Favorable prognosis in adults; seen in RUNX 1 -RUNX 1 T 1 younger adults overall 9896/3 AML with inv (16)(p 13 q 22) Favorable prognosis with standard therapy or t(16; 16)(p 13; q 22), CEFBMYH 11 9871/3 AML with t(9; 11)(p 22; q 23); MLLT 3 -M-LL Unusual age distribution-seen in ¾ of children >1 yr old; very poor prognosis 9897/3 AML with t(6; 9)(p 23; q 34); DEK-NUP 214 High rate of FLT 3 -ITD mutations which is conveys a poor prognosis 9865/3 AML with inv (3)(q 21 q 26: 2) or t(3; 3)(q 21; 126: 2); RPN 1 EV 11 Commonly presents with increase megakayocytes, many morphologically abnormal 9869/3 AML (magakayoblastic) with Rare; sometimes presents as a mass and t(1; 22)(p 13; q 13); mimics sarcoma RBN 15 -MKL 1 9911/3

AML with Recurrent Genetic Abnormalities • Codes that do not need a 20% blast for definitive diagnosis: – AML with t(8; 21)(q 22; q 22), RUNX 1 -RUNX 1 T 1 – AML with inv(16)(p 13 q 22) or t(16)(p 13; q 22), CEFB-MYH 11 – APL with t(15; 17)(q(22; 112), PML-RARA

Treatment Planning • Standard Treatment – Induction chemotherapy 7+3 regimen • Cytarabine plus an anthracycline – Post Remission Treatment: based on Risk Stratification • Hematopoeitic Stem Cell Transplant • Consolidation chemotherapy: HDAC: high-dose Ara-C, cytarabine-one dose every 28 days for 3 -4 doses

Factor Age Favorable Unfavorable <50 years >60 years Leukemia De novo secondary WBC <25, 000 >100, 000 FAB subtype M 3, M 4 M 0, M 5, M 6, M 7 Cytogenetics T(15; 17), t(8; 21) in(16) normal: RUNX 1 -RUNX 1 T 1; Abnormalities of chr. 5, 7, multiple, >3 abnormalities; 11 q 23, t (6; 9); inv 3 Extramedullary disease Absent present Auer rods Present Absent Phenotype CD 34 - CD 34+; CD 56+ Other Nucleophosmin 1 (NPM-1), CEBPA FLT-3 mutation; KIT mutations

The Older Adult with AML • Factors that contribute to poor outcomes: – – – – Poorer performance status Higher incidence of multidrug resistance Lower percentage of favorable cytogenteics Higher percentage of unfavorable cytogenetics Higher treatment-related morbidity and mortality Higher incidence of treatment-resistant disease Lower complete remission rates, shorter remission durations, shorter median overall survival – Fewer opportunities for allo HCT

Treating the Older Adult with AML • Favorable or intermediate risk patients – Induction therapy 7+3 regimen • Unfavorable risk or patients with significant comorbidities – Supportive care • Transfusions, antibiotics, low-dose chemotherapy

AML Coding Tips • Histology Coding – Enter a provisional diagnosis, NOS, until a more specific diagnosis is made – There is no time limit • Treatment Coding – 1 st course treatment includes all remission-inducing and remission-maintaining therapies – Treatment can span a year or more – Supportive care is not coded as treatment

Acute Promyelocytic Leukemia • Biologically and clinically distinct variant of AML – FAB classification of AML-M 3 – WHO classification: APL with t(15; 17)(q 24. 1; q 21. 1); PML-RARa (9866/3) • Incidence: 5 -8% of all AML cases • Age-predominately adults in mid-life

Clinical Manifestations of APL • Presents as a clinical emergency with a high rate of early mortality – Often due to hemorrhage from coagulopathy • Presenting symptoms typically secondary to pancytopenia: anemia, neutropenia, thrombocytopenia – Easy fatigability – Infections – Bleeding • Bleeding for APL patients may be secondary to DIC

APL and DIC • APL is often manifested by Disseminated Intravascular Coagulation – DIC is a cascade of bleeding and micro clotting that leads to a depletion of clotting factors and platelets • Ultimately it leads to hemorrhage in various sites in the body – This is a medical emergency • Untreated it may lead to pulmonary or cerebral hemorrhage and possible death due to hemorrhage – May be present at diagnosis or at initiation of cytotoxic therapy – Induction of therapy improves condition

Diagnostic Criteria of APL • Suspected by the characteristic morphology of the leukemic cells • Presence of severe coagulopathy • The Non-granular form of APL presents with leukopenia • Diagnosis is confirmed by testing for the characteristic PML-RARA fusion gene or associated chromosomal translocation – Usually by FISH testing

Treatment of APL • May span 1 -2 years total – Remission induction – Consolidation – Maintenance

Treatment of APL • Induction – Treatment should be initiated as soon as APL is suspected • Median survival of <1 month without treatment – ATRA-Retinoic Acid • Mechanism: induces tumor cell differentiation and maturation • If the patient is found to have another type of leukemia, ATRA will be d/c’d – Plus chemotherapy • An anthracycline and possibly Cytarabine

Treatment of APL • Consolidation: – Arsenic Trioxide followed by a combination of an antrhacycline plus ATRA • Maintenance – ATRA daily for one year

Coding Tips for APL • APL is considered a distinct variant of AML and should be coded as such • Per the SEER Antineoplastic Drug Database – ATRA is coded • Code under “Other Treatment” and code as (1), Cancer treatment not otherwise assigned – Arsenic Trioxide is not a coded drug at this time • The precise mechanism of action has not been fully determined

MYELODYSPLASTIC SYNDROMES

Myelodysplastic Syndromes • Group of heterogeneous malignant hematopoietic stem cell disorders • Characteristics – Dysplastic and ineffective blood cell proliferation – Variable risk of transformation to acute leukemia • May occur de novo or after exposure to mutagenic therapy (radiation, chemotherapy)

MDS: Epidemiology and Risk Factors Age distribution Median age at diagnosis: 65 Gender Male predominance Environmental risk factors: Chemical exposure: benzene, radiation, tobacco, chemotherapy drugs) Genetic risk factors trisomy 21, Fanconi anemia, Bloom syndrome Comorbid conditions as risk factors Benign hematologic diseases (paroxysmal nocturnal hemagloinburia, congenital nuetropenia)

Clinical Presentation of MDS • Non-specific signs and symptoms – Many patients are asymptomatic • Most common presenting signs are from a cytopenia – Anemia is the most common cytopenia – Infection is a less common presentation

Diagnostic Criteria of MDS • Clinical evaluation with pathologic evaluation of peripheral blood and bone marrow – Unexplained changes at least one lineage that quantifies as a cytopenia – Morphologic dysplasia on visual inspection – Blast forms <20% of total cells

Types of MDS Refractory anemia <5% blasts; only anemia 9980/3 Refractory neutropenia >10% dysplastic neutrophils 9991/3 Refractory thrombocytopenia >10% dysplastic megakaryocytes; cytogenetic studies helpful 9992/3 Refractory anemia with ring sideroblasts >15% ring sideroblasts in BM; <5% blasts in peripheral blood; 9982/3 Refractory anemia with multilineage dysplasia Bi-cytopenia/pancytopenia and dysplastic changes in 2 or more myeloid lines 9985/3 Refractory anemia with excess blasts Multiple types based on blast percentage from 1 -19% 9983/3 MDS associated with isolated del 5 q Associated with specific genetic abnormality 9986/3
, -7, trisomy 8, del (20")
MDS: Chromosomal Abnormalities • Most common: – Del(5 q), -7, trisomy 8, del (20 q) and loss of the Y chromosome • Single or multiple chromosomal changes may be present at the time of diagnosis • Chromosomal changes may occur during the course of the disease Do not change the coding of MDS once the initial diagnosis has been made

MDS: 5 q-Syndrome • Distinctive profile – Median age at diagnosis is 65 -70 years – Female predominance of 7: 3 • Typical presentation – refractory macrocytic anemia – normal or elevated platelets – absence of neutropenia • Benign course of disease – Projected median survival is 63 months – Likelihood of transformation to AML is low

Classification of MDS-IPSS-R • International Prognostic Scoring System. Revised – Calculates prognosis based on: • • • Bone marrow blast percentage Karyotype (very good, intermediate, very poor) Hemoglobin Platelets Absolute Neutrophil count – Stratifies patients into 5 risk categories • Survival and AML evolution
 Median time to")
IPSS-R Scoring System Risk Group IPSS-R Score Median Overall Survival (years) Median time to 25 percent AML evolution (years) Very low <1. 5 8. 8 >14. 5 Low >1. 5 to 3. 0 5. 3 10. 8 Intermediate >3 to 4. 5 3. 0 3. 2 High >4. 5 to 6 1. 4 Very High >6 0. 8 0. 7

Treatment of MDS • Asymptomatic disease: watch and wait – When patients develop transfusion* requirement or recurrent infections, this may herald the need for treatment

Treatment of MDS • Clinical trials • Supportive care, antibiotics, and transfusions • Low intensity chemotherapy – Hematopoietic growth factors, azacitidine, decitabine • May improve QOL, but not curative • High intensity therapy – Combination chemotherapy and HCT

Treatment of MDS • Most treatment regimens continue until disease progression – Disease progression demonstrated by • Worsening cytopenias • Increase in the percentage of bone marrow blasts • Progression to a more advanced MDS FAB subtype

MDS Coding Tips • Keep the initial diagnostic code of MDS once established – If transformation to AML occurs, this is a new primary • Often transfusions and growth factors are initial forms of treatment – Neither of these are currently coded for MDS
 • Occurs after treatment – Chemotherapy, radiation, stem cell")
Therapy Related Myeloid Neoplasm (9920/3) • Occurs after treatment – Chemotherapy, radiation, stem cell transplant or bone marrow transplant, • Peripheral blood and bone marrow are principle sites • May present as either t-MDS or t-AML – WHO has one group – Must have MD statement

MULTIPLE MYELOMA: PLASMA CELL MYELOMA

Multiple Myeloma Disease characteristic Clinical manifestation Neoplastic proliferation of plasma cells in bone marrow Bone pain; anemia, hypercalcemia; pathologic fractures Plasma cell production of monoclonal immunoglobulin Increased total serum and/or urine concentration of M protein Plasma cell infiltration of organs; organ Kidney damage from immunoglobulin light chains

Multiple Myeloma: Epidemiology Race African Americans: Caucasian 2 to 3 times higher Gender Slightly more frequent in men than women Age Average at diagnosis: 66

Diagnostic Criteria • Clonal bone marrow plasma cells > 10% • Or biopsy proven bony or soft tissue plasmacytoma • PLUS one of the following: – Presence of related organ or tissue impairment: • • Increased Calcium level Renal insufficiency Anemia Bone lesions – Presence of a biomarker associated with end-organ damage
")
Symptoms of MM Symptom Cause Calcium levels increase Product of osteoclast activating factors (hypercalcemia) Renal dysfunction Damage from light chain infiltration; M globulin infiltration and damage to kidney Anemia Secondary to bone marrow replacement of normal hematopoeitic tissue by tumor and disruption of bone marrow microenvironment Bone lesions, osteolytic The hallmark sign of MM; due to increased osteoclast activity with suppression of osteoblasts
 – Protein electrophoresis of")
Diagnosis • Laboratory Testing – Protein electrophoresis of serum (SPEP) – Protein electrophoresis of aliquot of urine (UPEP) – Serum free light chain assay (FLC) – Urinalysis • Myeloma cast uropathy – Peripheral smear – Bone marrow exam • IHC, cytogenetics, free light chain assay

Diagnosis • Radiographic Studies – Plain radiographs of the humeri and femoral bones • Key component to pt evaluation – CT, MRI and PET/CT scan – Bone scan is not a preferred method of evaluation

Related Conditions • • • MGUS Smoldering MM* Non-secretory MM* Plasma cell leukemia* Solitary plasmacytoma Solitary extramedullary plasmacytoma

Staging MM • Durie-Salmon Staging – M-protein levels – Calcium levels – Bone damage – Hemoglobin • International Staging System – Beta-2 microglobulin – Serum albumin

Transplant Eligibility • Patients are considered ineligible for one of the following – Age > 77 – Bilirubin >2. 0 mg/d. L – ECOG performance status 3 or 4 – New York Heart Association functional status Class III or IV

Treatment for MM High Risk 4 cycles of VRd or VCd Assess Response/collect Stem cells Autologous HCT if eligible Bortezomib-based maintenance Intermediate Risk Standard Risk Eligible for transplant? 4 cycles of VCd Autologous HCT if eligible Bortezomib-based maintenance Yes 4 cycles of Rd or VCd Autologous HCT Lenolidomide-based maintenance Rajkumar, S. Overview of the management of multiple myeloma. In: Up. To. Date, Post TW (Ed) Up. To. Date, Waltham, MA (accessed 2/12/15. ) No Rd until progression

Multiple Myeloma Coding Tips • Dexamethasone is often part of the induction therapy • Maintenance therapy can go on for an extended period of time – Maintenance would not be considered subsequent treatment if it was planned

Summary • Coding histology should be as specific as possible for all these patients – Often a final diagnosis takes many weeks • Treatment for many hematologic malignancies can span a year or more – Treatment should be considered subsequent if it has not been considered as part of the initial treatment plan – Treatment would be considered subsequent if the patient showed signs of disease progression

How can you become an expert in abstracting any kind of bone marrow cancer?

http: //seer. cancer. gov/seertools/hemelymph/

http: //seer. cancer. gov/seertools/hemelymph/

http: //seer. cancer. gov/seertools/hemelymph/

http: //seer. cancer. gov/seertools/hemelymph/
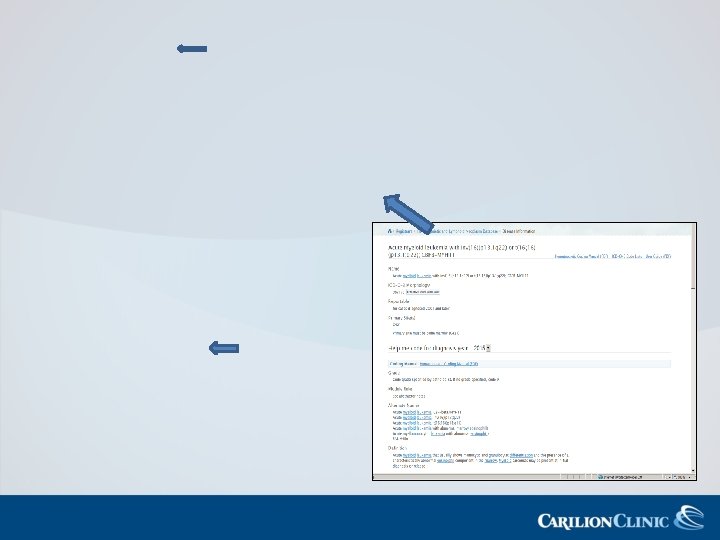
(p 13. 1 q 22) 9871/3 http: //seer.")
AML, NOS, 9861/3 AML with inv (16)(p 13. 1 q 22) 9871/3 http: //seer. cancer. gov/seertools/hemelymph/

“Any fool can know. The point is to understand. ” Albert Einstein

References • • • • Acute Myeloid Leukemia, NCCN Clinical Practice Guidelines in Oncology, version 1. 2015, (2015)NCCN. org. (Accessed 2/24/15) Aster, J and Stone, R. Clinical manifestations and diagnosis of the myelodysplastic syndromes. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/12/15. ) www. cancedr. org/cancer/multiplemyeloma/detailedguide/multiple-myeloma-staging (Accessed 3/7/15) Facility Oncology Registry Data Standards. (2015) American College of Surgeons Estey, E and Schrier S. Prognosis of the myelodysplastic syndromes in adults. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/12/15. ) Larson, R. Induction therapy for acute myeloid leukemia in younger adults. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/06/15. ) Larson, R. , Initial Treatment of acute promyelocytic leukemia in adults. In Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 9/23/15) Larson, R. Post-remission therapy for acute myeloid leukemia in younger adults. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/06/15. ) Larson, R. and Klepin, H. Pretreatment evaluation and prognosis of acute myeloid leukemia in older adults. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/06/15. ) Larson, R. Thereapy-related myeloid neoplasms: Acute myeloid leukemia and myelodysplastic syndrome Larson, R. Treatment of acute myeloid leukemia in older adults. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/06/15. ) Larson, R. and Anastasi, J et. Clinical manifestations, pathologic features and diagnosis of acute promyelocytic leukemia in adults; In: Up. To. Date, Waltham, MA. (Accessed 9/23/15) Multiple Myeloma NCCN Clinical Practice Guidelines in Oncology, v 3. 2015. (2015) NCCN. org. (Accessed 2/24/15) Myelodysplastic Syndromes, NCCN Clinical Practice Guidelines in Oncology, v. 2. 2015, (2015) NCCN. org. (Accessed 2/24/15. )

References • • • Rakjumar, S. Clinical features, laboratory manifestations, and diagnosis of multiple myeloma. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/12/15. ) Rajkumar, S. Overview of the management of multiple myeloma. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/12/15. ) Rajkumar, S. Pathobiology of multiple myeloma. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/12/15. ) Ruhl. , J and Adamo, M et al. Hemaotpoietic and Lymphoid Neoplasm Coding Manual (2015), NCI SEER. Schiffer, C and Anastasi, J. Clinical manifestations, pathologic features, and diagnosis of acute myeloid leukemia. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/06/15. ) Schiffer, C. Prognosis of acute myeloid leukemia. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/06/15. ) Stock, W and Thirman, M. Molecular genetics of acute promyelocytic leukemia. In Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA (Accessed 9/23/15). Stock, W and Thirman, M. Pathogenesis of acute myeloid leukemia. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/12/15. ) Zhang, Y. and Le Beau, M. Cytogenetics in acute myeloid leukemia. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/06/15. ) Zhang, Y and Beau, M. Cytogenetics and molecular genetics of myelodysplastic syndromes. In: Up. To. Date, Post TW (ED), Up. To. Date, Waltham, MA. (Accessed 2/12/15. )
- Slides: 65