Cancers in children hematologic malignancies leukemia lymphoma prof






 • acute lymphoblastic leukemia • Acute myeloblastic leukemia")
 • precursor B-cell ALL - 80 -85% • mature B-cell")
 • Auer rods • Staining: myeloperoxidase (+), monocyte-associated esterases (+),")

 petechiae(50%) lymphadenopathy (35%)")




 • symptoms and signs are similar to ALL • acute")
 • in contrast to ALL, therapy for AML is of")

")
 • Childhood NHL • disseminated, • diffuse not nodular, • high-grade")
 • lymph node biopsy/resection,")
 Small noncleaved (Burkitt")






: • weight loss, night sweats, rash and")
- Slides: 32
Cancers in children: hematologic malignancies leukemia, lymphoma prof. Mariusz Wysocki, Katedra i Klinika Pediatrii, Hematologii i Onkologii Collegium Medicum Bydgoszcz UMK Toruń
Cancers in children: prevalence
Cancers in children and age
Cancer Associated syndrome Leukemia Trisomy 21, Bloom syndrome, Fanconi anaemia, ataxia telangiectasia, neurofibromatosis, Kostmann syndrome, Klinefelter syndrome, Li-Fraumeni syndrome, Diamond-Blackfan anaemia, Noonan syndrome CNS Tumours Neurofibromatosis, tuberous sclerosis, Li-Fraumeni syndrome, von Hippel-Lindau syndrome Lymphoma Immunodeficiency disorders Wilms tumour Denys-Drash syndrome, Beckwith-Weidermann syndrome, WAGR syndrome Rhabdomyosarcoma Li-Fraumeni syndrome
Risk of development • siblings of children with leukemia - increased risk of developing leukemia (approximately 2 – 4 fold above the childhood population • risk increases for twin siblings (up to 25% for monozygotic twins) • familial leukemia • environmental factors: ionizing radiation, chemotherapy agents
Leukemia: pathogenesis • While the cause of leukaemia remains unknown, the prevailing theory for leukemic development is that a mutant stem cell, capable of indefinite renewal, gives rise to abnormal proliferation of lymphoblasts (ALL) or myeloblasts (AML) in the bone marrow. These cells occupy the marrow space, leading to reduced numbers of normal haematopoietic cells, resulting ultimately in pancytopenia. Secondary involvement of the reticuloendothelial system (leading to lymphadenopathy and hepatosplenomegaly), bone, joints and rarely CNS, testes and skin can occur. A two-step pathogenesis for ALL (Greaves' hypothesis) has been suggested, with the initial event, occurring during fetal life, driving clonal expansion and a second trigger occurring during childhood, possibly resulting from viral stimuli of cellular proliferation. This theory stems from evidence that a significant proportion of children presenting with ALL have molecular evidence of leukaemic clones identified retrospectively at birth on newborn screening cards.
Leukemia: classification • acute leukemia (AL) • acute lymphoblastic leukemia • Acute myeloblastic leukemia • Chronic leukemia - 95% - 80% - 15% - 5% In ALL, presentation peaks at age 2 -5 years, whereas there is no peak in AML. Certain constitutional genotypes can predispose a child to the development of acute leukemia. Patients with Down syndrome, Fanconi anemia, Bloom syndrome, ataxia-telangiectasia, Wiskott-Aldrich syndrome, and neurofibromatosis type 1 all have an increased risk of acute leukemia. Siblings of children with leukemia are also at increased risk of developing leukemia (approximately two- to fourfold above the childhood
Acute lymphoblastic leukemia (ALL) • precursor B-cell ALL - 80 -85% • mature B-cell ALL - 2 - 3% • T-cell ALL - 10 -15% • staining: myeloperoxidase (-), monocyte-associated esterases (-); Periodic acid –Schiff (PAS)(-) • Classification: • French/American/British (FAB): • 1 of 3 morphological subclasses Leukaemia can be further classified on the basis of morphological characteristics, immunobiology and cytogenetic or molecular markers. Immunophenotype as determined by flow cytometric detection of cell surface antigens is commonly used to differentiate distinct ALL categories into: precursor B-cell ALL (early pre-B, pre. B, transitional pre-B) - 80 -85% ; mature B-cell ALL - 2 -3% ; T-cell ALL - 10 -15%.
Acute myeloblastic Leukemia (AML) • Auer rods • Staining: myeloperoxidase (+), monocyte-associated esterases (+), PAS () • Classification: • French/American/British (FAB): • one of eight morphological subclasses • genetic features of leukaemic cells In AML, characteristic morphological and cytochemical features include the presence of Auer rods as well as positive staining for myeloperoxidase and monocyte-associated esterases. Classification into one of eight morphological subclasses using the French/American/British (FAB) system is possible. In addition to morphology and immunophenotype, genetic features of leukaemic cells can provide diagnostic and prognostic information.
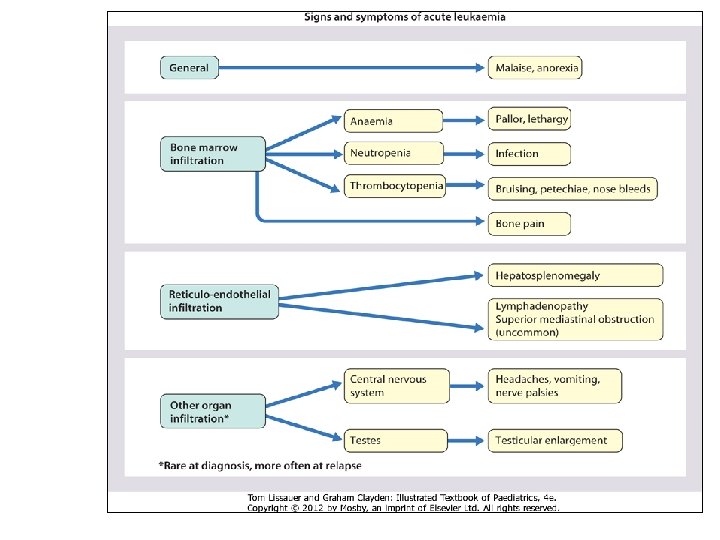
ALL: clinical presentation 3 – 4 week prodrome • pallor, • increased bruising or bleeding, • lethargy, anorexia, • recurrent infection or fevers, • bone pain or reluctance to walk The clinical presentation of ALL can be quite variable; however, most children will present with a 3 -4 -week prodrome that may include pallor, increased bruising or bleeding, lethargy, anorexia, recurrent infection or fevers, anorexia, bone pain or reluctance to walk
ALL: clinical presentation – physical examination • • • pallor (80%) petechiae(50%) lymphadenopathy (35%) hepatomegaly or splenomegaly (50 -60%) rarely, skin infiltration (chloroma) and testicular infiltration (usually presents as a painless swelling). The clinical presentation of ALL can be quite variable; however, most children will present with a 3 -4 -week prodrome that may include pallor, increased bruising or bleeding, lethargy, anorexia, recurrent infection or fevers, anorexia, bone pain or reluctance to walk. Signs and symptoms of acute leukemias are related to the infiltration of leukemic cells into normal tissues, resulting in either bone marrow failure (anemia, neutropenia, thrombocytopenia) or specific tissue infiltration (lymph nodes, liver, spleen, brain, bone, skin, gingiva, testes). Common presenting symptoms are fever, pallor, petechiae or ecchymoses, lethargy, malaise, anorexia, and bone or joint pain. Physical examination frequently reveals lymphadenopathy and hepatosplenomegaly. Symptomatic central nervous system (CNS) involvement is rare at the time of presentation. The testes are a common extramedullary site for ALL; a painless enlargement of one or both testes may be seen. Patients with T-cell ALL are frequently older males (8 -10 years), and often have high white blood cell (WBC) counts, anterior mediastinal masses, bulky disease with cervical lymphadenopathy, hepatosplenomegaly, and CNS involvement.
Leukemia: lab tests • full blood count • low haemoglobin, • thrombocytopenia • WBC and circulating leukaemic blast cells • bone marrow examination • chest X ray • cytometry • genetics Full blood count - in most but not all children, the blood count is abnormal, with low haemoglobin, thrombocytopenia and evidence of circulating leukaemic blast cells. Bone marrow examination is essential to confirm the diagnosis and to identify immunological and cytogenetic characteristics which give useful prognostic information. Chest X ray is required to identify a mediastinal mass characteristic of T-cell disease.
Leukemia: WBC • ALL • < 10 × 109/l • 10 -50 × 109/l • > 100 × 109/l • AML - 25% pts - 50% pts - 25% pts • > 100 × 109/l - 21% - 10% The white blood cell count (WCC) is frequently elevated at diagnosis (leucocytosis), with a presenting WCC below 10 × 109/l in 25%, 10 -50 × 109/l in 50% and above 50 × 109/l in 25% of patients. A bone marrow aspirate and biopsy (trephine) are the gold standard diagnostic tests and will show replacement of normal haematopoiesis by leukaemic cells. A lumbar puncture is also done during the staging work-up with approximately 5 -10% of patients showing leukaemic spread to the cerebrospinal fluid (CSF). T-cell leukaemia, more common in older boys, presents with a mediastinal mass in 50% that can result in life-threatening airway compression and obstruction of the superior vena cava. 30% of patients with T-cell ALL present with a leucocyte count greater than 100 × 109/l and there is a higher incidence of CNS disease.
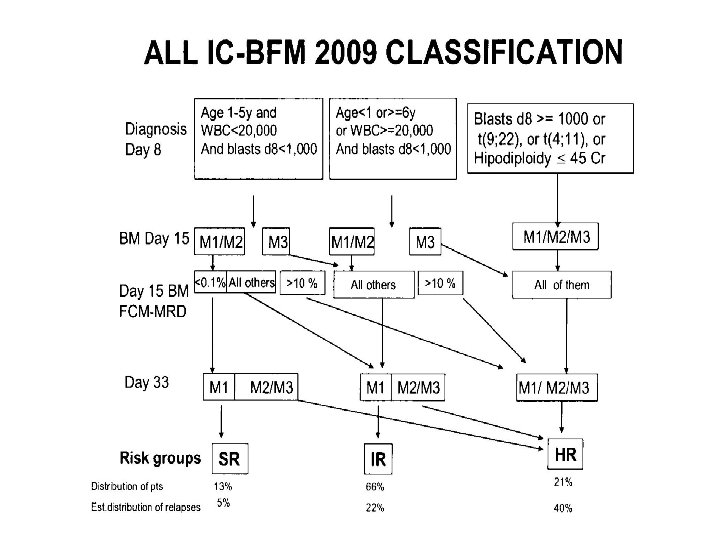
Risk group Clinical features Molecular/genetic features Low risk Age 2 -10 years, WCC <50 × 109/1 DNA index >1. 16 Not T-cell phenotype Absence of: t(9; 22) BCR-abl No central nervous system or testicular disease t(4; 11) MLL/AF 4 Rapid response to induction therapy t(1; 19) MLL rearrangement t(12; 21) TEL/AML 1 High risk and very high Induction failure risk Age < 12 months Poor prednisone response High MRD levels t(9; 22), t(4; 11) MLL rearrangements
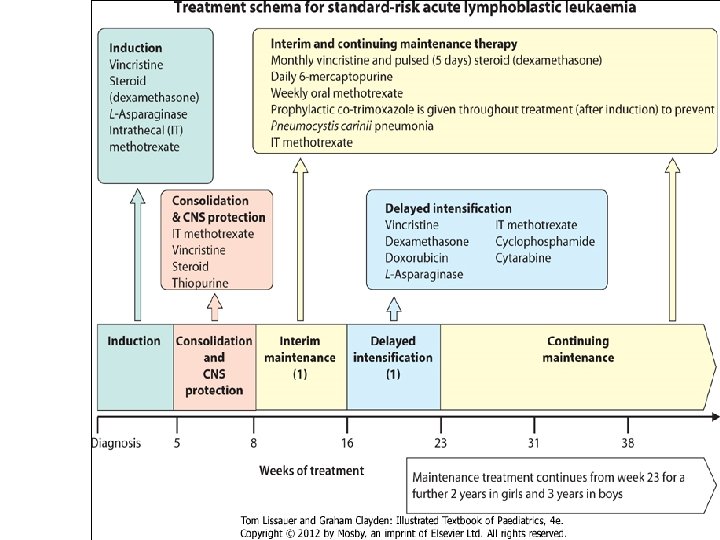
• Induction: Four weeks of combination chemotherapy is given and current induction treatment schedules achieve remission rates of 95%. Intensification A block of intensive chemotherapy is given to consolidate remission. This improves cure rates but at the expense of increased toxicity. Central nervous system Cytotoxic drugs penetrate poorly into the CNS. As leukaemic cells in this site may survive effective systemic treatment, additional treatment with intrathecal chemotherapy is used to prevent CNS relapse. Continuing therapy Chemotherapy of modest intensity is continued over a relatively long period of time, up to 3 years from diagnosis. Co-trimoxazole prophylaxis is given routinely to prevent Pneumocystis carinii pneumonia. Treatment of relapse High-dose chemotherapy, usually with total body irradiation (TBI) and bone marrow transplantation, is used as an alternative to conventional chemotherapy after a relapse.
Acute myeloblastic leukemia (AML) • symptoms and signs are similar to ALL • acute promyelocytic leukaemia (APML) • serious haemorrhage or disseminated intravascular coagulation • acute monoblastic or myelomonoblastic leukaemia • skin infiltration (chloroma) or gum hypertrophy AML accounts for 20% of acute leukaemia. Presenting symptoms and signs are similar to ALL and can include pallor, bleeding, fever, anorexia, malaise and bone pain. Certain subtypes of AML have more distinctive presenting clinical features. Acute promyelocytic leukaemia (APML) can present with serious haemorrhage or disseminated intravascular coagulation, whereas acute monoblastic or myelomonoblastic leukaemia may present with skin infiltration (chloroma) or gum hypertrophy. CNS leukaemia is diagnosed in 5 -15% of patients. Like ALL, the differential diagnosis can include infection, juvenile rheumatoid arthritis, idiopathic thrombocytic purpura, aplastic anaemia and osteomyelitis.
Acute myeloblastic leukemia (AML) • in contrast to ALL, therapy for AML is of shorter duration and more intensive • several cycles of extremely intensive myelosuppressive chemotherapy • BMT • low-dose continuation therapy
In contrast to ALL, therapy for AML is of shorter duration and more intensive, often requiring frequent hospital admissions with aggressive supportive care, including blood products and antimicrobials during lengthy periods of marrow suppression. he treatment of AML is quite different from that of ALL because nonmyelosuppressive drugs (vincristine, prednisone, and asparaginase) are not effective. Several cycles of extremely intensive myelosuppressive chemotherapy are necessary to cure childhood AML; there is little evidence that low-dose continuation therapy is helpful in AML (with the exception of acute promyelocytic leukemia). Induction therapy for AML usually consists of cytarabine, daunomycin, and etoposide (or 6 -thioguanine). Induction is most effective for long-term outcome when two courses of drugs are given consecutively (1 to 2 weeks apart) regardless of blood counts, as opposed to waiting for counts to recover after the first course. If a patient has an HLAmatched sibling donor, most experts recommend a stem cell transplantation in the first remission, Overall, the outlook for patients with AML is less optimistic, with survival rates reported of 50 -70%.
Lymphomas • 10% of childhood cancers • two basic types: • non-Hodgkin lymphoma (NHL) • Hodgkin disease • prevalence: boys > girls • adenopathy – biopsy: • persistent adenopathy (>2 -3 weeks) • site of adenopathy (e. g. supraclavicular) • character (firm, >1 -2 cm) Lymphomas, accounting for approximately 10% of childhood cancers, are third most common form of malignancy in childhood. There are two basic types: non-Hodgkin lymphoma (NHL) and Hodgkin disease. Both are more common in boys than in girls. Lymphomas are malignancies of the cells of the immune system and can be divided into Hodgkin and non-Hodgkin lymphoma (NHL). NHL is more common in childhood, while Hodgkin lymphoma is seen more frequently in adolescence. Although lymphadenopathy attributable to an infectious aetiology is more common in childhood, any child with persistent adenopathy (>2 -3 weeks) should be considered for a biopsy. Site of adenopathy (e. g. supraclavicular) or character (firm, >1 -2 cm) may indicate the need for earlier biopsy.
Non-Hodgkin lymphoma (NHL) • Childhood NHL • disseminated, • diffuse not nodular, • high-grade immature T- or B-cell lineage • frequent spread to extranodal sites, marrow and CNS • Adult NHL • predominantly nodal involvement • low-grade malignancy Childhood NHL has quite different features from its adult counterpart. Childhood NHL is more often disseminated, diffuse not nodular, high-grade immature T- or B-cell lineage with frequent spread to extranodal sites, marrow and CNS. In contrast, NHL occurring in adulthood is usually a low-grade malignancy with predominantly nodal involvement. Clinical and pathological staging is achieved with organ imaging (computed tomography (CT) of
NHL: staging • CT: chest/abdomen/pelvis, • positron emission tomography (PET) • lymph node biopsy/resection, • bone marrow aspirate and biopsy (trephine) • CSF examination • when > 25% of bone marrow is involved, disease is classified as T- or Bcell ALL (CT) of chest/abdomen/pelvis, positron emission tomography (PET) scan or gallium scan), lymph node biopsy/resection, bone marrow aspirate and biopsy (trephine) and CSF examination. When more than 25% of bone marrow is involved, disease is classified as T- or B-cell ALL
NHL classification Histologic Category Immunophenotype Usual Primary Site Most Common Translocation(s) Small noncleaved (Burkitt lymphoma) Mature B-cell (surface immunoglobulin present) Abdomen (sporadic form) Head and neck (endemic form) t(8; 14)(q 24; q 32) t(2; 8)(p 11; q 24) t(8; 22)(q 24; q 11) Lymphoblastic T-cell (rarely pre-Bcell) Neck and/or anterior mediastinum Many Large cell T-cell, B-cell, or indeterminate Lymph nodes, skin, soft tissue, bone t(2; 5)(p 23; q 35)
Almost all cases of NHL in childhood are diffuse, are highly malignant, and show little differentiation. NHL has three histologic subtypes: small noncleaved cell (Burkitt lymphoma), lymphoblastic, and large cell. For simplicity, these may be regarded as B cell, T cell, and large cell (which may be of either B-cell or T-cell origin).
NHL: clinical features • mediastinal, primary of T-cell - 25% • acute superior vena cava syndrome • airway obstruction (stridor, cough, pleural effusion) • patient: preteen or early teenage males • abdominal NHL, B-cell - 35 -40% • local tumour (intussusception) • massive diffuse abdominal disease (ascites)
T-cell malignancies may present as acute lymphoblastic leukaemia or non. Hodgkin lymphoma, with both being characterised by a mediastinal mass with varying degrees of bone marrow infiltration. The mediastinal mass may cause superior vena caval obstruction. B-cell malignancies present more commonly as non-Hodgkin lymphoma, with localised lymph node disease usually in the head and neck or abdomen. Abdominal disease presents with pain from intestinal obstruction, a palpable mass or even intussusception in cases with involvement of the ileum. A mediastinal primary of T-cell immunophenotype accounts for 25% of NHL and often presents with acute superior vena caval and/or airway obstruction (a medical emergency) producing stridor and cough, usually with an associated pleural effusion and characteristically occurring in preteen or early teenage males. Diagnosis, immunophenotyping and cytogenetics may be made on pleural aspirate, suprasternal or supraclavicular node biopsy, or rarely on direct biopsy of the mediastinal mass. Abdominal lymphoma accounts for 3540%, is of B-cell immunophenotype and characteristically presents as either local tumour causing intussusception and readily removable, or massive diffuse abdominal disease, often with ascites. The later is often associated with uricacid-induced nephropathy or tumour lysis syndrome. Release of uric acid, potassium and phosphate from rapidly growing tumours, particularly following commencement of chemotherapy, can result in significant renal impairment and life-threatening electrolyte disturbances (hyperkalaemia, hyperphosphataemia, hypocalcaemia)
NHL: treatment • pathological diagnosis and staging • multiagent chemotherapy ± auto BMT • prognosis - cure rate: • stages I and II • stages III and IV - > 90% pts - 0 -80% pts Following pathological diagnosis and staging, multiagent chemotherapy is initiated; the intensity and duration depends upon stage and immunophenotype. Stages I and II have a more than 90% cure rate and stages III and IV a 70 -80% cure rate.
Hodgkin disease • prevalence: boys > girls, • rare before the age of 5 years • progressively increasing incidence in adolescents • viral aetiology? - Epstein-Barr virus • painless progressive swelling of lymph nodes • dissemination to spleen, liver, lungs, bones and bone marrow can occur
Hodgkin disease, more common in boys than girls, is rare before the age of 5 years, with a progressively increasing incidence in adolescents. A viral aetiology is suspected, with the genome of Epstein-Barr virus identified in some Hodgkin cells; however, the significance of this is not clear. Classically presents with painless lymphadenopathy, most frequently in the neck. Lymph nodes are much larger and firmer than the benign lymphadenopathy commonly seen in young children. The lymph nodes may cause airways obstruction The clinical history is often long (several months) and systemic symptoms (sweating, pruritus, weight loss and fever - the so-called 'B' symptoms) are uncommon, even in more advanced disease. A painless progressive swelling of lymph nodes (above the diaphragm in two-thirds of patients) is the most common clinical presentation. Dissemination to spleen, liver, lungs, bones and bone marrow can occur. Constitutional symptoms, including weight loss, night sweats, rash and fever, occur in one-third of patients. Open biopsy confirms the diagnosis and pathological staging with CT chest/abdo/pelvis, gallium or PET scan and marrow aspirate and trephine completes the workup. Chemotherapy is the mainstay of treatment, with radiotherapy having a supplemental role in patients with massive mediastinal involvement. Cure rates are excellent, with survival greater than 90%. Emphasis on cure without cost has become paramount in this disease, with a shift to therapy combinations that allow for preservation of fertility, reduction in rates of secondary cancers (associated with the use of radiation and etoposide) and reduced longer-term organ morbidity (e. g. lung toxicity with bleomycin, cardiomyopathy with anthracyclines) without compromising cure rates.
Hodgkin disease • constitutional symptoms (1/3 pts): • weight loss, night sweats, rash and fever • diagnosis and staging: • open biopsy (Reed-Sternberg cells) • CT chest/abdo/pelvis, PET scan • marrow aspirate and trephine • treatment: chemotherapy ± radiotherapy • cure rates: survival > 90% pts The pathologic hallmark of Hodgkin disease is the identification of Reed. Sternberg cells. Histopathologic subtypes in childhood Hodgkin disease are similar to those in adults; 10% to 15% have lymphocyte predominant, 50% to 60% have nodular sclerosis, 30% have mixed cellularity, and less than 5% have lymphocyte depletion. All subtypes are responsive to treatment. Staging of Hodgkin disease is according to the Ann Arbor system. Chemotherapy is the mainstay of treatment, with radiotherapy having a supplemental role in patients with massive mediastinal involvement. Cure rates are excellent, with survival greater than 90%. Combination chemotherapy with or without radiotherapy. Positron emission tomography (PET) scanning is used in the UK to monitor treatment response and guide further management Overall, about 80% of all patients can be cured. Even with disseminated disease, about 60% can be cured.