Building Phylogenies DistanceBased Methods Methods Distancebased Parsimony Maximum

Building Phylogenies Distance-Based Methods

Methods • Distance-based • Parsimony • Maximum likelihood

Distance Matrices a b c d 0 6 7 14 a a 0 3 10 b b 0 9 c 0 d c d 0 1 2 34 5 6 7 8 Distance matrix is additive if there is a tree that fits it exactly

Ultrametric Matrices a b c d 0 2 6 10 a a 0 6 10 b b 0 10 c 0 d c d 0 1 2 34 5 Additive + molecular clock assumption

Methods • • Fitch - Margoliash UPGMA Neighbor-joining Many others

Least squares trees • Minimize over all trees • Choice of weights wij : – Uniform: wij 1 – Fitch-Margoliash: wij 1/Dij 2 – Others. . .
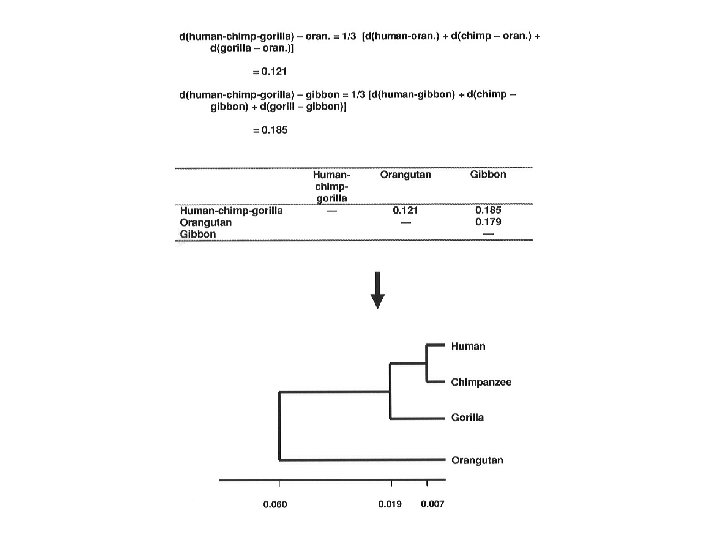
 immunological distances")
Sarich's (1969) immunological distances

Least squares tree for Sarich’s data

Clustering Methods • E. g. , UPGMA and Neighbor-Joining • A cluster is a set of taxa • Interspecies distances translate into intercluster distances • Clusters are repeatedly merged – “Closest” clusters merged first – Distances are recomputed after merging

UPGMA • Unweighted pair group method using arithmetic averages • The distance between clusters Ci and Cj is • After merging Ci and Cj to create cluster Ck define distance from k to every other cluster r as

UPGMA: Initialization 1. Assign each sequence i to its own cluster Ci 2. Define one leaf (tip) of tree for each sequence and place it at height 0

UPGMA: Iteration Repeat until only two clusters remain: 1. Choose the two clusters i and j with smallest Dij 2. Create a new cluster k, where Ck = Ci Cj 3. Compute Dkr for all r. 4. Define a new node k with children i and j, and place it at height Dij /2. 5. Add k to the current clusters and delete i and j Let i and j be the remaining clusters. Place root at height Dij /2

UPGMA Example



UPGMA tree for Sarich’s data

A pitfall of UPGMA • The algorithm produces an ultrametric tree: the distance from the root to any leaf is the same • UPGMA assumes a constant molecular clock: all species accumulate mutations (evolve) at the same rate.

UPGMA fails when molecular clock assumption doesn’t hold
 • Idea:")
Neighbor Joining • Saitou and Nei, Molecular Biology and Evolution 4 (1987) • Idea: Find a pair of leaves that are close to each other but far from other leaves – Implicitly finds a pair of neighboring leaves • Advantages: – Works well for additive and other nonadditive matrices – Does not have the molecular clock assumption

Long branches must be handled carefully! 0. 1 0. 4 and are closer to each other than to or . Obvious approach produces incorrect clusters!

Compensating for long edges Introduce “correction terms” Average dist. to other taxa “Corrected” distances: Distances are reduced for pairs that are far away from all other species: They may be close to each other.

Neighbor-joining Repeat the following until only two leaves remain: 1. Choose i, j such that Dij ui uj is minimum 2. Define a new leaf k whose distances to i and j are 3. Compute the distance from k to every other leaf r 4. Delete i and j Connect the 2 remaining leaves by a branch of length Dij

NJ tree for Sarich’s data

Computing distance matrices • Based on sequence alignment • Various possibilities: – Distance = average number of differences – Try different PAM matrices; distance = index of matrix that gives highest score – Feng and Doolitle: Based on alignment scores – roughly ratio to max possible score (see text) • Read, e. g. , PHYLIP documentation: http: //evolution. genetics. washing ton. edu/phylip/general. html

Distance correction • The amount of evolutionary change is not linearly related to time • Over a long period of time, a series of substitutions may bring us back to where we started • Percentage difference may underestimate evolutionary time

Jukes-Cantor Model

Correcting for multiple substitutions in the JC model

Many other models!
- Slides: 29