BTEC 564 Protein Biotechnology GTU Fall 2017 Isil

BTEC 564 Protein Biotechnology GTU, Fall 2017 Isil AKSAN KURNAZ

A little updated syllabus: 1. Biochemical characteristics of proteins. Protein synthesis. The overview of the differences between the eukaryotic and prokaryotic protein synthesis 2. Protein folding, 3 D structure formation. Chaperons. Folding problems, folding diseases. 3. Protein sorting and targeting 4. The posttranslational modification of proteins and their analysis 5. Proteins as biological effectors (immunoglobulins, protein hormones, enzymes …. ) Lecture based on, but not limited to: (see syllabus for official textbooks) 6. Recombinant protein technology 7. Heterologous expression systems 8. Protein purification and analysis 9. Production of human therapeutic proteins and enzymes 10. Large scale industrial protein production, GLP and GMP 11. Protein improvement: Protein engineering and design of more effective proteins 12. Protein improvement: Synthetic biology http: //www. google. com. tr/url? sa=t&rct=j&q=&esrc=s&source=web&cd=8&cad=rja&uact=8&ved=0 ah. UKEwigst 7 xrz. RAh. UMIMAKHWBa. Cd. YQFgh. TMAc&url=http%3 A%2 F%2 Fwww. tankonyvtar. hu%2 Fen%2 Ftartalom%2 Ftamop 4 25%2 F 0011_1 A_Proteinbiotech_en_pdf%2 FProteinbiotech_EN. pdf&usg=AFQj. CNHMIly. Va. Hk_Xok. HMZoi 2 y. OBv 1 QXg&sig 2=i. E 32 zrx. IIw. GUa. SXDIs. Cj. Ag

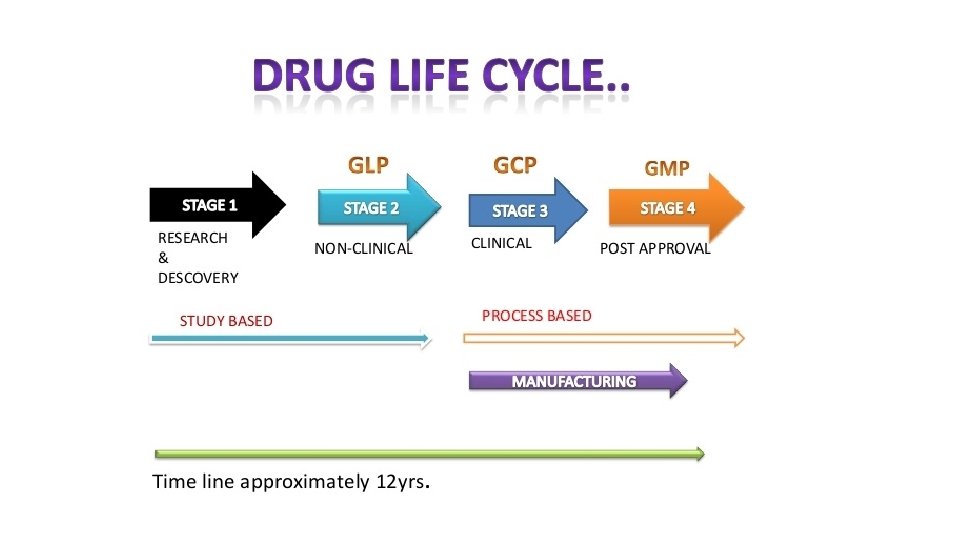
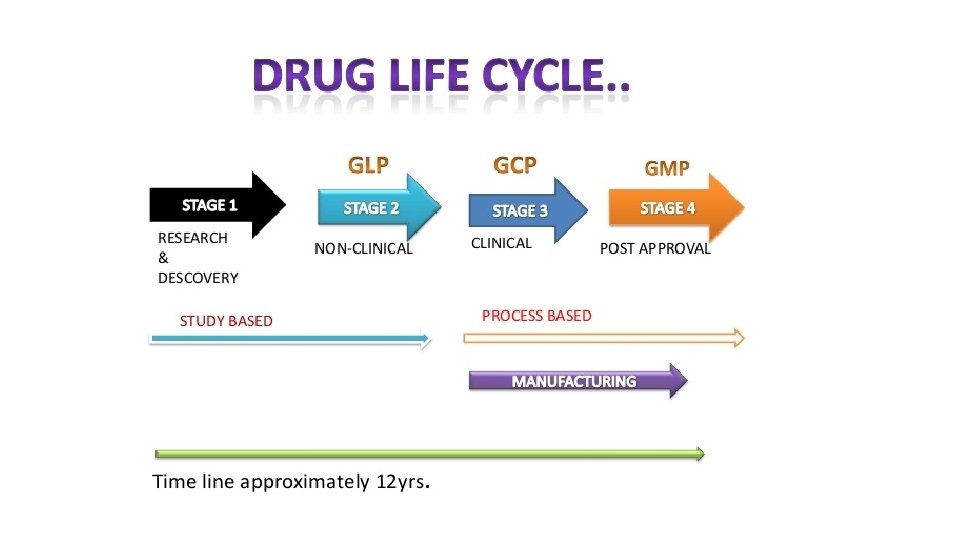
10. Large scale industrial protein production, GLP and GMP

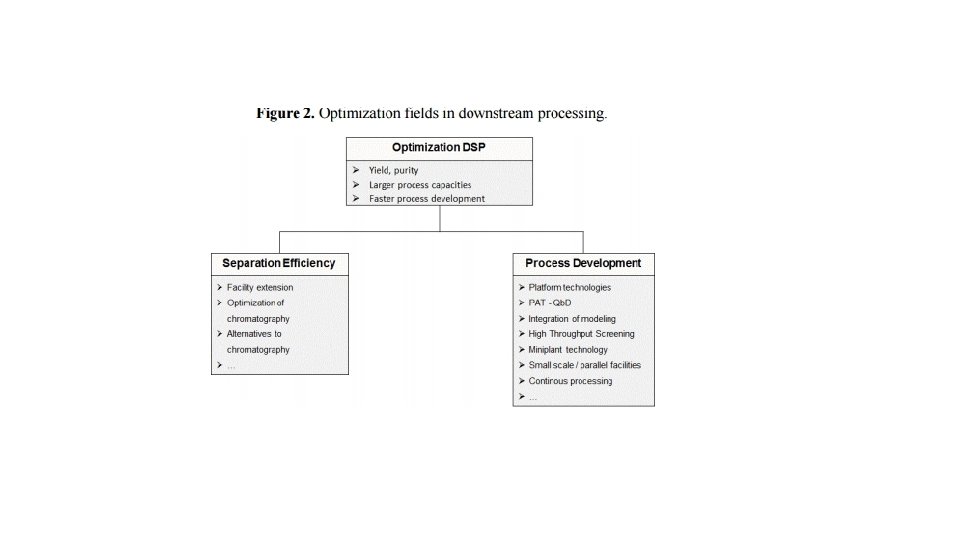
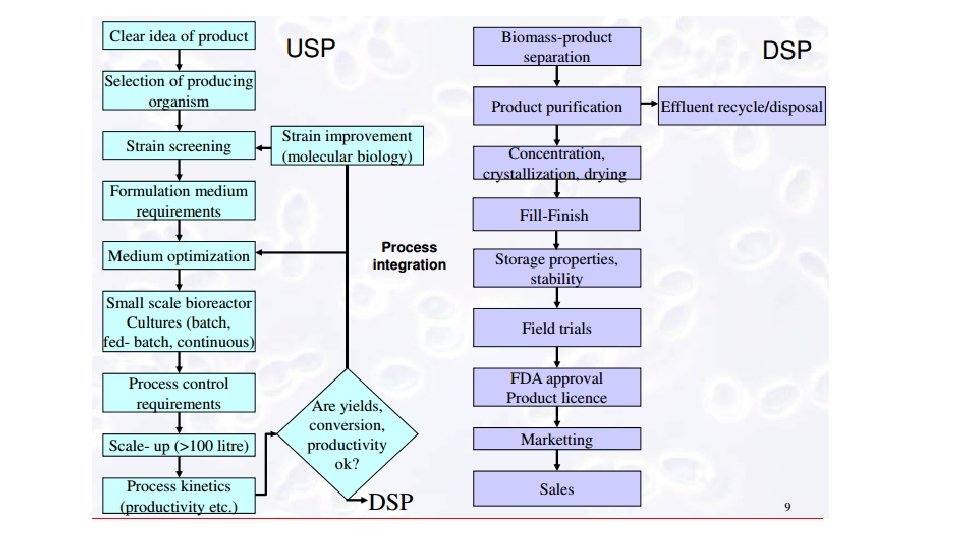
Scaling up protein purification difficulties with scaling up lab methods that work on small scale may not always be adaptable to large scale production changes in purification process can invalidate earlier small scale clinical studies Why would it be necessary to seek FDA approval again for an approved bioengineered protein if the process changes when it is scaled up? FDA must always approve the process for producing the bioengineered protein so if the process changes then FDA must then approve the new process used to do large scale production.

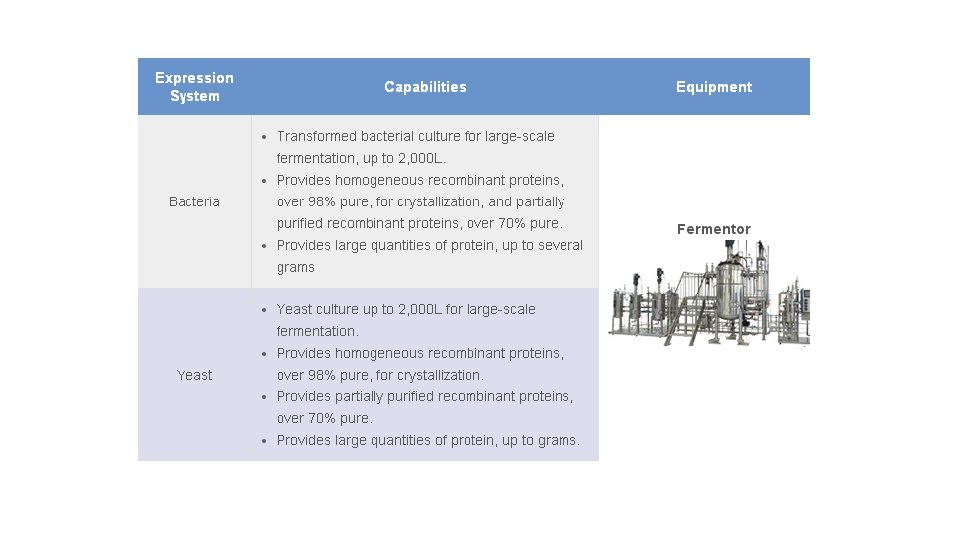
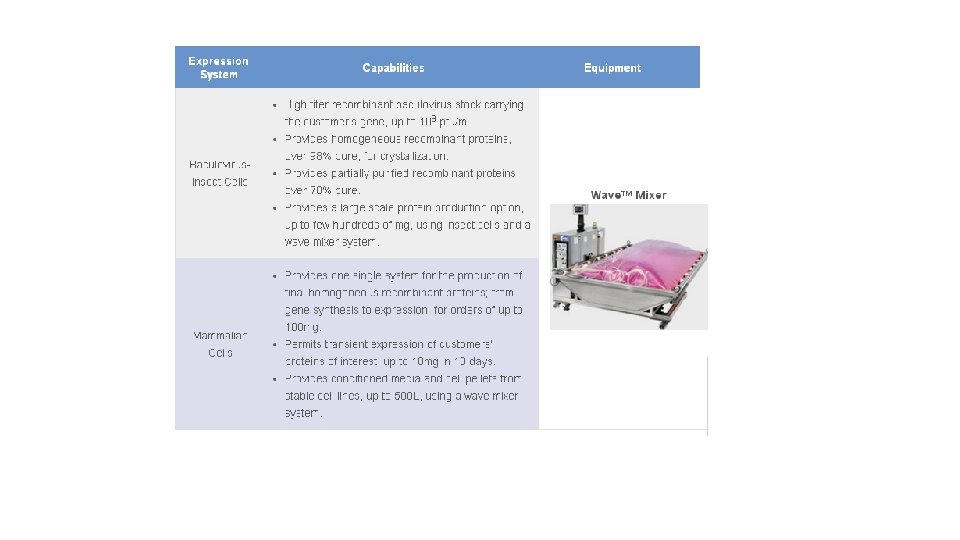

Industrial fermentation is the intentional use of fermentation by microorganisms such as bacteria and fungi as well as eukaryotic cells like CHO cells and insect cells, to make products useful to humans. Fermented products have applications as food as well as in general industry. Some commodity chemicals, such as acetic acid, citric acid, and ethanol are made by fermentation. The rate of fermentation depends on the concentration of microorganisms, cellular components, and enzymes as well as temperature, p. H and for aerobic fermentation oxygen. Product recovery frequently involves the concentration of the dilute solution. Nearly all commercially produced enzymes, such as lipase or invertase, are made by fermentation with genetically modified organisms.

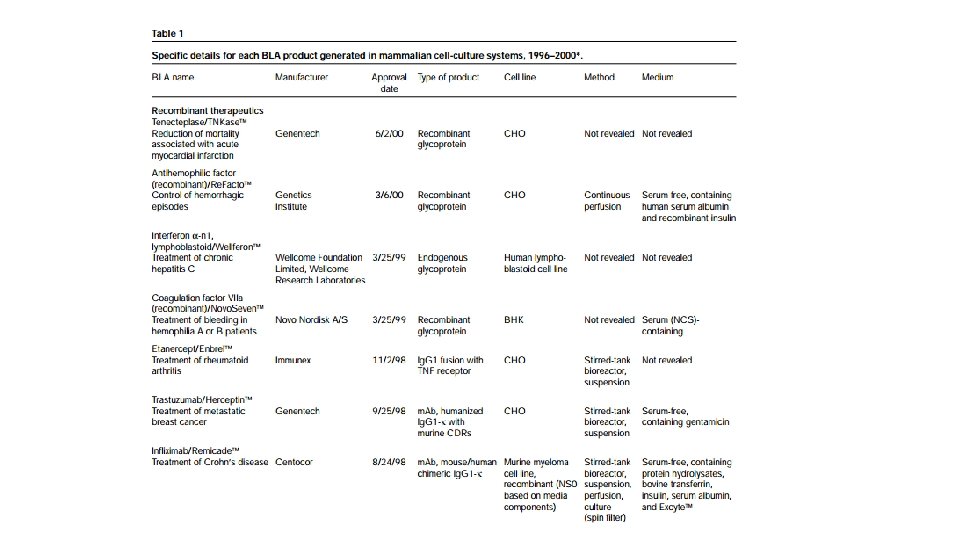
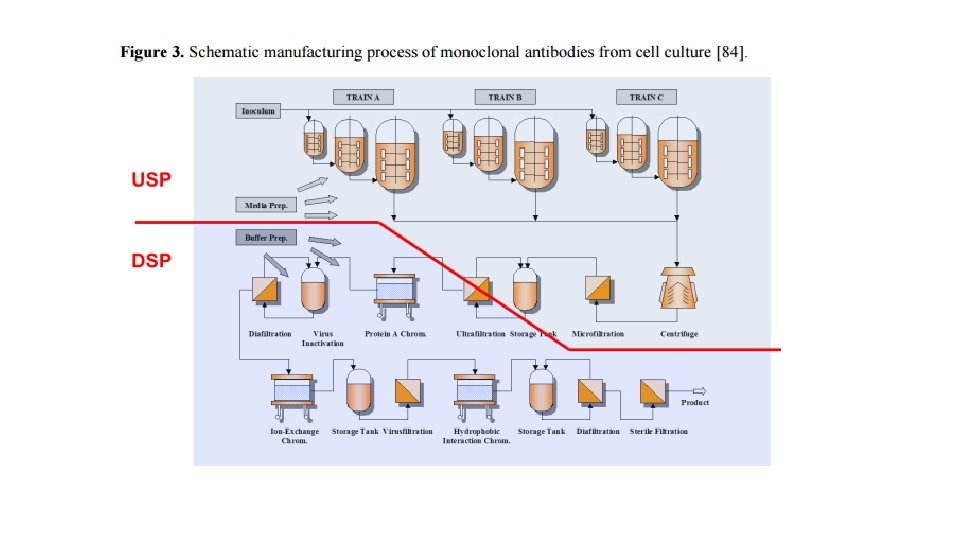
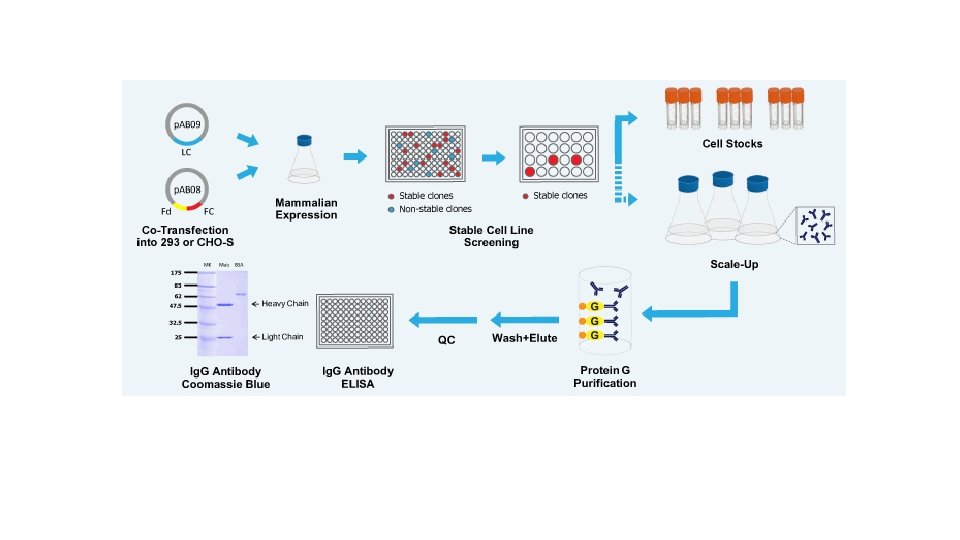
At an industrial scale, the manufacture of a protein product is often divided into upstream and downstream processing. Commercialized proteins are most commonly produced as follows: Therapeutic proteins, Diagnostic/analytical proteins, and Industrial (bulk) proteins. Microbial cell fermentation has a long history in the production of various biological products of commercial significance, including ethanol, antibiotics, organic acids and proteins, and can be easily adapted to large-scale purification of recombinant proteins.

 or")
Genetically engineered bacteria or cells can be grown in large scale fermenters (anaerobic) or bioreactors (aerobic) Computers monitor air in bioreactors keeping oxygen levels and temperature ideal for cell growth When phase of cell growth is highest- the bacteria promoter must be activated to stimulate foreign gene expression Must activate gene in recombinant organism after the organism has completed synthesizing important natural proteins needed for its metabolism

http: //www. mdpi. com/2306 -5354/1/4/188/htm

http: //www. mdpi. com/2306 -5354/1/4/188/htm

http: //www. mdpi. com/2306 -5354/1/4/188/htm

http: //www. mdpi. com/2306 -5354/1/4/188/htm










http: //www. who. int/tdr/publications/documents/glp-handbook. pdf
 research arena, the phrase good laboratory practice or GLP specifically")
In the experimental (non-clinical) research arena, the phrase good laboratory practice or GLP specifically refers to a quality system of management controls for research laboratories and organizations to ensure the uniformity, consistency, reliability, reproducibility, quality, and integrity of chemical (including pharmaceuticals) non-clinical safety tests; from physio-chemical properties through acute to chronic toxicity tests. GLP was first introduced in New Zealand Denmark in 1972, and later in the US in 1978 in response to the Industrial Bio. Test Labs scandal. It was followed a few years later by the Organization for Economic Co-operation and Development (OECD) Principles of GLP in 1992; the OECD has since helped promulgate GLP to many countries. GLP applies to non-clinical studies conducted for the assessment of the safety or efficacy of chemicals (including pharmaceuticals) to man, animals and the environment. GLP, a data quality system, should not be confused with standards for laboratory safety appropriate gloves, glasses & clothing to handle lab materials safely.

GLP is a quality system concerned with the organizational process and conditions under which non-clinical health and environmental safety studies are planned, performed, monitored, recorded, archived and reported. GLP principles include: Equipment, reagents and Materials Organization and Personnel Management-Responsibilities Sponsor-Responsibilities Study Director-Responsibilities Principal Investigator-Responsibilities Study Personnel-Responsibilities Test systems Physical/Chemical Biological Quality assurance program Quality Assurance Personnel Facilities Test System Facilities for Test and Reference Items Test & Reference items Standard operating procedures Performance of Study Plan Conduct of Study Reporting of results Archival - Storage of Records and Reports

http: //www. who. int/tdr/publications/documents/glp-handbook. pdf


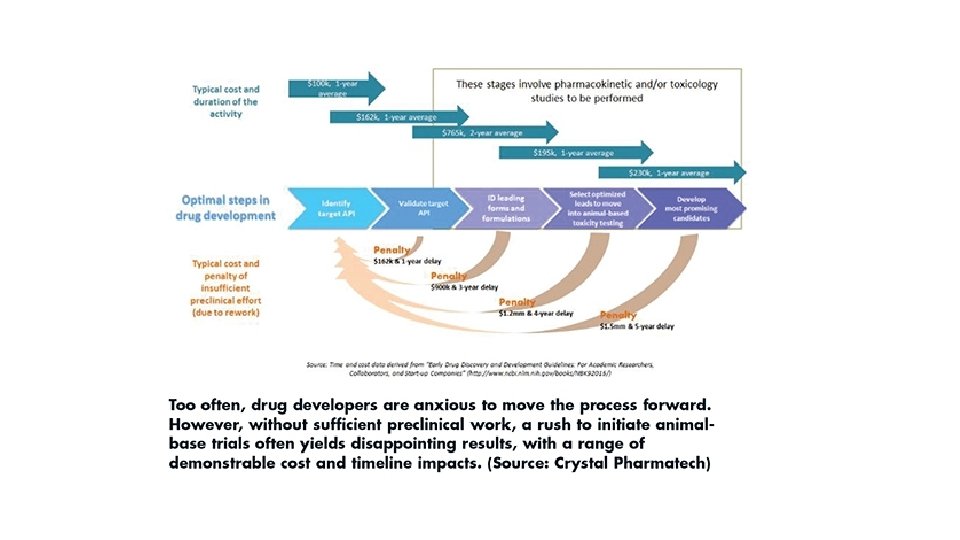
 or ADME (Absorption, Distribution, Metabolism, and Elimination) tests •")
DMPK (Drug Metabolism and Pharmacokinetics) or ADME (Absorption, Distribution, Metabolism, and Elimination) tests • Absorption – How much of the drug is absorbed and how quickly? (bioavailability) • Distribution- Where is the drug distributed within the body? What is the rate and extent of the distribution? • Metabolism- How fast is the drug metabolized? What is the mechanism of action? What metabolite is formed and is it active or toxic? • Elimination- How is the drug excreted and how quickly? • Toxicity-Does this drug have a toxic effect to body systems or organs?
 has produced several guidance documents for industry such")
The Food and Drug Administration (FDA) has produced several guidance documents for industry such as Safety Testing of Drug Metabolites, In Vitro Metabolism and Transporter-Mediated Drug-Drug Interactions Studies, Clinical Drug Interaction Studies — Study Design, Data Analysis, Clinical Implications, and Title 21 part 58 Good Laboratory Practices for Nonclinical Laboratory Studies to provide instruction and to ensure that best practices are employed when evaluating the safety and efficacy of a drug candidate. The underlying goal and end-game for all ADME/Tox studies is to better understand a compound’s metabolite-mediated toxicity and safety profile to make a concrete decision on whether the compound can progress to late stage preclinical and clinical studies to enable filing for an Investigational New Drug (IND), New Drug Agreement (NDA), or a Biologics Licensing Agreement (BLA). https: //www. thermofisher. com/blog/connectedlab/the-role-of-adme-toxicology-studies-in-drug-discovery-development/ https: //www. fda. gov/drugs/types-applications/investigational-new-drug-ind-application

During discovery, as well as in late stage preclinical and non-clinical studies, in vivo studies are conducted to evaluate pharmacokinetic (PK) properties. In vivo PK studies are conducted with Association of Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited animals such as mice and rats, whereas non- human primates are employed to generate PK data to evaluate properties such as drug clearance, bioavailability, exposure, half-life, and distribution volume. These studies include a mix of non-GLP and GLP toxicology studies. https: //www. thermofisher. com/blog/connectedlab/the-role-of-adme-toxicology-studies-in-drug-discovery-development/

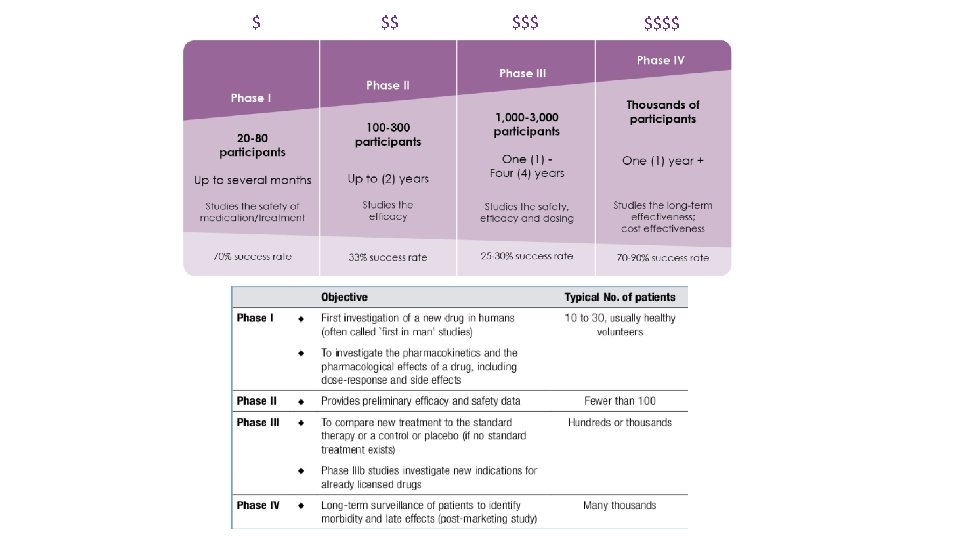
A clinical trial is a carefully designed study which tests the benefits and risks of a specific medical treatment or intervention, such as a new drug or a behavior change (e. g. , diet). Once researchers have completed a rigorous screening and preclinical testing process, the company files an Investigational New Drug (IND) application with the U. S. Food and Drug Administration (FDA). This application allows the investigational medicine to be tested in human volunteers in clinical trials. Every clinical trial is led by a principal investigator, who is usually a doctor, along with a team of nurses and others researchers. The FDA requires a multi-phase clinical trials process to be completed before deciding if the medicine under investigation is safe and effective for a broader patient population. Usually, the number of human volunteers in the trial increases as the treatment moves through these phases, which is why innovative medicines cannot be developed without the help of volunteers who participate in clinical trials.


 is an international quality standard that is provided by ICH,")
Good clinical practice (GCP) is an international quality standard that is provided by ICH, an international body that defines a set of standards, which governments can then transpose into regulations for clinical trials involving human subjects. A similar guideline for clinical trials of medical devices is the international standard ISO 14155, which is valid in the European Union as a harmonized standard. These standards for clinical trials are sometimes referred to as ICH-GCP or ISO-GCP to differentiate between the two and the lowest grade of recommendation in clinical guidelines. GCP follows the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) of GCP guidelines. GCP enforces tight guidelines on ethical aspects of a clinical study. High standards are required in terms of comprehensive documentation for the clinical protocol, record keeping, training, and facilities, including computers and software. Quality assurance and inspections ensure that these standards are achieved. GCP aims to ensure that the studies are scientifically authentic and that the clinical properties of the investigational product are properly documented. GCP guidelines include protection of human rights for the subjects and volunteers in a clinical trial. It also provides assurance of the safety and efficacy of the newly developed compounds. GCP guidelines include standards on how clinical trials should be conducted, define the roles and responsibilities of clinical trial sponsors, clinical research investigators, and monitors. In the pharmaceutical industry monitors are often called clinical research associates.

GCP is defined as « a standard for the design, conduct, performance, monitoring, auditing, recording, analysis and reporting of clinical trials that provides assurance that: - the data and reported results are credible and accurate, and that - the rights, integrity and confidentiality of the trial subjects are protected» Hence, there are 3 pillars that should be carefully monitored: Patient Data Study • Rights, safety and well-being of subjects prevail over interests of society • Individuals involved in trial should be qualified by education, training and experience to perform his/her duties • Information recorded, handled and stored to allow accurate reporting, interpretation and verification and confidentiality of subjects’ records • Trials should be scientifically sound and ethical in all aspects • Quality of every aspect of trial should be secured • Conducted acc to Helsinki declaration (’ 96) • Protocol should provide inclusion and exclusion criteria, monitoring and publication policy • Investigator/sponsor should cconsider all relevant guidance
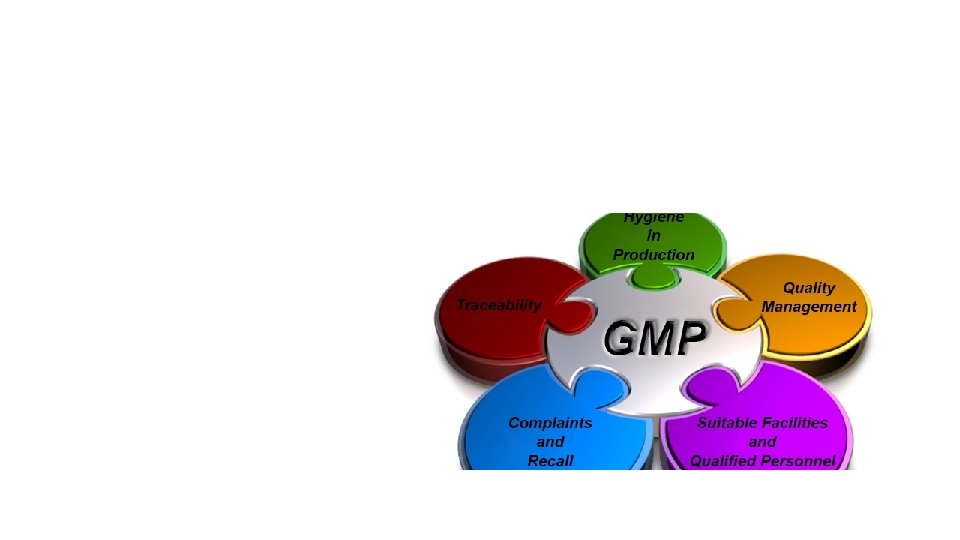
 are the practices required in order to conform to the")
Good manufacturing practices (GMP) are the practices required in order to conform to the guidelines recommended by agencies that control authorization and licensing for manufacture and sale of food, drug products, and active pharmaceutical products. These guidelines provide minimum requirements that a pharmaceutical or a food product manufacturer must meet to assure that the products are of high quality and do not pose any risk to the consumer or public.

All guidelines follow a few basic principles: 1. Manufacturing facilities must maintain a clean and hygienic manufacturing area. 2. Controlled environmental conditions in order to prevent cross contamination of food or drug product from adulterants that may render the product unsafe for human consumption. 3. Manufacturing processes are clearly defined and controlled. All critical processes are validated to ensure consistency and compliance with specifications. 4. Manufacturing processes are controlled, and any changes to the process are evaluated. Changes that affect the quality of the drug are validated as necessary. 5. Instructions and procedures are written in clear and unambiguous language. (Good Documentation Practices) 6. Operators are trained to carry out and document procedures. 7. Cross contamination with unlabelled major allergens is prevented. 8. Records are made, manually or by instruments, during manufacture that demonstrate that all the steps required by the defined procedures and instructions were in fact taken and that the quantity and quality of the food or drug was as expected. Deviations are investigated and documented. 9. Records of manufacture (including distribution) that enable the complete history of a batch to be traced are retained in a comprehensible and accessible form. 10. The distribution of the food or drugs minimizes any risk to their quality. 11. A system is available for recalling any batch from sale or supply. 12. Complaints about marketed products are examined, the causes of quality defects are investigated, and appropriate measures are taken with respect to the defective products and to prevent recurrence. http: //www. who. int/biologicals/vaccines/good_manufacturing_practice/en/


http: //www. who. int/biologicals/areas/vaccines/Annex_2_WHO_Good_manufacturing_practices_for_biological_products. pdf? ua=1


http: //www. who. int/medicines/areas/quality_safety/quality_assurance/TRS 986 annex 2. pdf? ua=1


CGMP refers to the Current Good Manufacturing Practice regulations enforced by the US Food and Drug Administration (FDA). CGMPs provide for systems that assure proper design, monitoring, and control of manufacturing processes and facilities. Adherence to the CGMP regulations assures the identity, strength, quality, and purity of drug products by requiring that manufacturers of medications adequately control manufacturing operations. This includes establishing strong quality management systems, obtaining appropriate quality raw materials, establishing robust operating procedures, detecting and investigating product quality deviations, and maintaining reliable testing laboratories. This formal system of controls at a pharmaceutical company, if adequately put into practice, helps to prevent instances of contamination, mix-ups, deviations, failures, and errors. This assures that drug products meet their quality standards.



- Slides: 60