Bone Marrow Failure Syndromes Brian Boulmay MD Or

Bone Marrow Failure Syndromes Brian Boulmay, MD

Or, It’s Fixing To Get Real Brian Boulmay, MD

Bone Marrow Failure • Ineffective marrow-poeisis is the final endpoint of many diseases. ▫ Congenital ▫ Acquired �Genetic �Environmental or iatrogenic causes • Congenital marrow failure can present at any age. ▫ Usually in childhood. ▫ Some exceptions • Marrow failure often presents with single lineage declines

Bone Marrow Failure • Early onset of marrow disorders can give a clue to genetic v. acquired syndromes • Dysmorphic body findings ▫ Some may present later in life: �dyskeratosis congenita • Genetic testing will be confirmatory

Inherited Syndromes • Inheritance patterns of inherited forms of marrow failure are variable: ▫ X linked ▫ Autosomal recessive ▫ Autosomal dominant • Not every inherited syndrome has a clear genetic etiology

Inherited Syndromes • Many of the inherited syndromes have an increased risk of acute leukemia and solid tumors. • Penetrance of a disorder may vary within a family.

Marrow morphology • Examination of marrow morphology in a patient with single or multi-lineage cytopenias can be very non-specific. • Often just see decreased marrow elements • Clinical correlates and genetic/molecular analysis is key.

• A six year old girl is seen for pancytopenia. Her history is significant for duodenal atresia, a solitary left kidney, and modest sensineural hearing loss. Labs show a Hct of 25%, a platelet count of 39 K and WBC of 2. 19 with lymphocyte predominance. What diagnostic test should you order? 1) Chromosome breakage analysis 2) Erythrocyte adenosine deaminase level 3) Pancreatic isoamylase level 4) T and B lymphocyte subsets 5) Telomere length analysis

• A six year old girl is seen for pancytopenia. Her history is significant for duodenal atresia, a solitary left kidney, and modest sensineural hearing loss. Labs show a Hct of 25%, a platelet count of 39 K and WBC of 2. 19 with lymphocyte predominance. What diagnostic test should you order? 1) Chromosome breakage analysis 2) Erythrocyte adenosine deaminase level 3) Pancreatic isoamylase level 4) T and B lymphocyte subsets 5) Telomere length analysis

Shwachman-Diamond Syndrome • Autosomal recessive. • Mutation of SBDS gene on Chr 7. ▫ The SBDS protein is involved with mitotic spindle stabilization ▫ Causes a proliferative defect • Short, pancreatic exocrine deficiency, neutropenia. ▫ May have severe anemia, thrombocytopenia ▫ Longterm increased risk of MDS/AML • Pancreatic isoamylase deficiency • Androgens, G-CSF are temporizing measure • SCT will correct the hematologic abnormality

Diamond-Blackfan Syndrome • Autosomal dominant ▫ Pure red cell aplasia • Mutation of the RPS 19 gene on Chr 19 ▫ Plurality of patients, 25% ▫ Involved in ribosome assembly • Considerable variation in presentation: ▫ Hydrops fetalis presentation in adulthood • Physical phenotype: “tow-colored hair, snub nose, wide set eyes, intelligent expression” ▫ Web neck, short stature, triphalangeal thumb Cathie Arch Dis Chil 1950
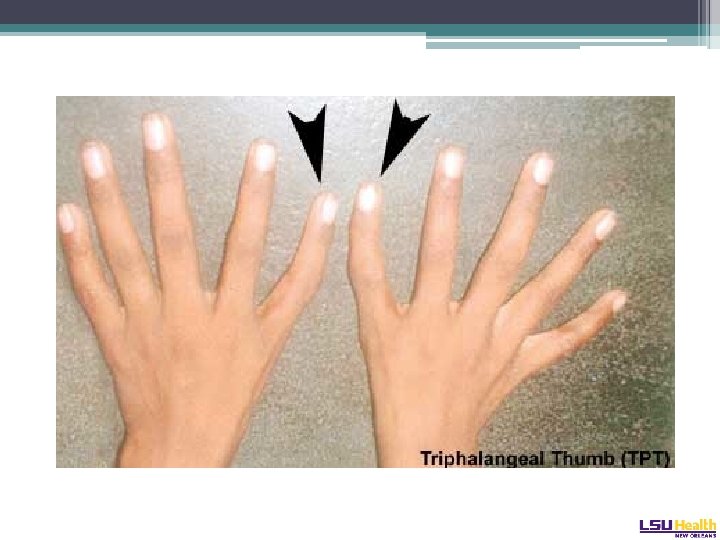

Diamond-Blackfan Syndrome • Marrow and peripheral blood triad: ▫ ▫ Anemia Reticulocytopenia Absence of red cell precursors in the marrow (Multilineage hypoplasia may develop in long term survivors) • Erythrocyte adenosine deaminase (ADA) levels high • Glucocorticosteroids are the classic therapy ▫ Not effective in all. Adverse effects in children. • SCT the definitive therapy.

• A 12 year old boy presents for evaluation of moderate thrombocytopenia. There is a family history of IPF in a grandfather at 50 years, MDS in his aunt. The boy’s skin is reticulated in appearance with some nail changes. Which test should you order to confirm the (obvious) abnormality? 1) 2) 3) 4) 5) Bone marrow evaluation Chromosome breakage analysis High resolution karyotype Skin biopsy Telomere length analysis

• A 12 year old boy presents for evaluation of moderate thrombocytopenia. There is a family history of IPF in a grandfather at 50 years, MDS in his aunt. The boy’s skin is reticulated in appearance with some nail changes. Which test should you order to confirm the (obvious) abnormality? 1) 2) 3) 4) 5) Bone marrow evaluation Chromosome breakage analysis High resolution karyotype Skin biopsy Telomere length analysis

Dyskeratosis congenita • X-linked, autosomal dominant or autosomal recessive. • Very rare • Mutations involve genes of telomere maintenance or the telomere complex* ▫ DKC 1 - X linked ▫ TERC- Dominant form ▫ TERT

Dyskeratosis congenita • Physical findings. The mucocutaneous triad: ▫ Leukoplakia ▫ Nail dystrophy ▫ Reticulated skin hyperpigmentation • Can be subtle and only appear later in life.

Dermopath. com, accessed 1/2014

Dyskeratosis congenita • Typically starts with anemia, with high MCV and Hgb F and thrombocytopenia • Progresses to pancytopenia • Median age at presentation is 16 years old • Diagnosis made by flow-FISH of telomere length or quant PCR of telomere DNA or leukocytes
 • Autosomal recessive inheritance •")
Fanconi Anemia • Very rare (1: 1, 000 births) • Autosomal recessive inheritance • Due to mutation in the FANC gene ▫ Protein products of this gene family protect against DNA cross linking. • Most patients present around 8 years old. ▫ Can present in adulthood.

Fanconi. co. za, accessed 12/2013

Fanconi Anemia • 60% of patients have a physical exam finding ▫ ▫ Absent radius, absent thumb Short Microcephaly, hypertelorism Skin pigment abnormalities (café au lait spots) • Confirmatory testing: lymphocyte chromosomal breakage on diepoxybutane exposure. ▫ Differentiates from aplastic anemia

Fanconi Anemia • Therapy: ▫ Androgenic steroids ▫ SCT • Risks: ▫ Increased risk for solid tumors, MDS and AML

Acquired single lineage marrow failure • • • Parvovirus B 19 Thymoma associated pure red cell aplasia Iron Deficiency Anemia of chronic inflammation Nutritional deficiencies ▫ B 12 ▫ Folate • Levamisole tainted cocaine

• A 17 year presents with one month of bruising. She feels very tired. Her medical and family histories are completely unremarkable. Her examination is normal, except for being pale. Her Hgb is 6 g/d. L, her WBC is 1. 0, platelet count is 7 K. Marrow shows:
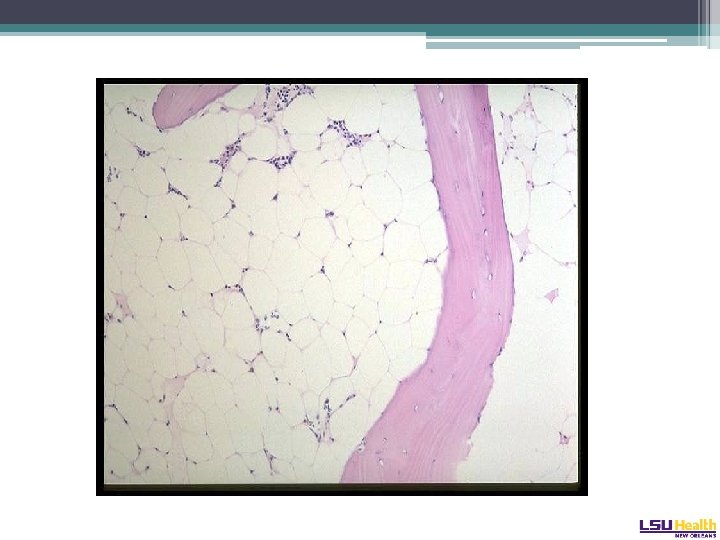

• The karyotype is normal. The fragility test to diepoxybutane is negative. How do you treat this patient?

• The karyotype is normal. The fragility test to diepoxybutane is negative. How do you treat this patient? 1) Horse ATG and Cy. A 2) Rabbit ATG and Cy. A 3) Alemtuzumab 4) HLA typing of an older sibling for allo. SCT 5) MUD SCT

• The karyotype is normal. The fragility test to diepoxybutane is negative. How do you treat this patient? 1) Horse ATG and Cy. A 2) Rabbit ATG and Cy. A 3) Alemtuzumab 4) HLA typing of an older sibling for allo. SCT 5) MUD SCT
 • Paroxysmal nocturnal hemoglobinuria • Myelodysplastic")
Acquired multilineage marrow syndromes • Aplastic anemia (autoimmune) • Paroxysmal nocturnal hemoglobinuria • Myelodysplastic syndrome • HIV • Hepatitis C • Bone marrow replacement ▫ Cancer ▫ Myelofibrosis B 12 def. Pregnancy SLE

Aplastic anemia • Frequency 1: 1, 000 • Occurs at any age • Progressive pancytopenia ▫ Often presents with severe pancytopenia • Needs to be differentiated from FA or DC, even in adults. ▫ Consider PNH in differential. • Rule out other marrow injuries ▫ HIV ▫ Hepatitis ▫ Drugs/radiation � Sulfas, seizure meds, HIV meds ▫ ETOH

Aplastic Anemia Cellularity <25% of normal Maturation usually normal Remnant cellularity usually lymphs, plasma cells Consider hypocellular MDS or PNH

Aplastic anemia • On marrow: ▫ No fibrosis ▫ No malignant cells ▫ Hemapoietic precursors do not appear dysplastic • Cytogenetics should be normal

Aplastic Anemia • A population of CD 55 - and CD 59 - cells may be found in patients with AA ▫ Maybe as high as 23% of lymphocytes • Does not mean patient has PNH ▫ Don’t treat as PNH • Predicts response to ATG/Cy. A Sugimori Blood 2006

Aplastic Anemia • Classification: ▫ Moderate aplastic anemia �Bone marrow cellularity <30 percent �Absence of severe pancytopenia �Depression of at least two of three blood elements below normal ▫ Severe aplastic anemia �A bone marrow biopsy showing <25 percent of normal cellularity, or �A bone marrow biopsy showing <50 percent normal �fewer than 30 percent of the cells are hematopoietic and at least two of the following are present: absolute reticulocyte count <40, 000/microliter; absolute neutrophil count (ANC) <500/microliter; or platelet count <20, 000/microliter. ▫ Very severe aplastic anemia �Criteria for severe aplastic anemia are met and the ANC is <200

Aplastic Anemia • Therapy: ▫ Combination of ATG /MP/ Cy. A results in higher response rates than with ATG/ MP alone � 70% v 41% at four months ▫ Lifetime relapse rates ultimately the same in both arms. Frickhofen Blood 2003

Aplastic Anemia

Aplastic Anemia

Aplastic Anemia • Horse ATG versus rabbit ATG: �Primary study endpoint was hematologic response at 6 months: � 68% versus 38% � 3 year OS favored horse ATG � 96% versus 76% Scheinberg NEJM 2011

Aplastic Anemia

Aplastic Anemia • Cy. A should be stopped at 6 months • 2 year taper after the usual 6 months can delay relapse, but does not appear to prevent it. Young Abs 2406 ASH 2011

Aplastic Anemia • Therapy guidelines: ▫ <20 with an HLA matched donor: transplant �Cat 2 C rec for MUD. ▫ 20 - 50 with an HLA matched donor: transplant ▫ >50: immunosuppression

Aplastic Anemia • Role of mimetics ▫ G-CSF and erythropoietin have no effect in AA ▫ Eltrombopag (ELT) is a TPO mimetic: �Triggers the mpl surface receptor. �Approved for use in ITP ▫ 24 patients treated with ELT with refractory AA � 40% response rate ▫ Mpl mutation now implicated in some familial AA Walne Hematologica 2011; Olnes NEJM 2012

• 35 year old woman treated 5 years previously for aplastic anemia with ATG/Cy. A. Her CBC normalized after therapy. However, her counts began to decline six months ago. Marrow biopsy shows hypercellularity and 5% blasts. The patient has no sibs. What is the best therapy at this time? 1) Repeat therapy with horse ATG and Cy. A 2) Change therapy to rabbit ATG and Cy. A 3) Alternative donor SCT 4) Induction chemotherapy 5) Androgens

• 35 year old woman treated 5 years previously for aplastic anemia with ATG/Cy. A. Her CBC normalized after therapy. However, her counts began to decline six months ago. Marrow biopsy shows hypercellularity and 5% blasts. The patient has no sibs. What is the best therapy at this time? 1) Repeat therapy with horse ATG and Cy. A 2) Change therapy to rabbit ATG and Cy. A 3) Alternative donor SCT 4) Induction chemotherapy 5) Androgens

• A 28 year old has transfusion dependent anemia for the past six months. Two years ago she developed the onset of periodic dark urine. Her Hgb was 7 g/d. L. Her WBC and platelet count are normal. Coombs is negative. Marrow shows 50% cellularity with no dysplasia; karyotype is normal. Flow demonstrates a loss of CD 55/CD 59. Diagnosis?

• A 28 year old has transfusion dependent anemia for the past six months. Two years ago she developed the onset of periodic dark urine. Her Hgb was 7 g/d. L. Her WBC and platelet count are normal. Coombs is negative. Marrow shows 50% cellularity with no dysplasia; karyotype is normal. Flow demonstrates a loss of CD 55/CD 59. • Treatment with eculizumab is begun. She receives therapy for two years, but does not become transfusion independent.

• A repeat marrow shows 15% cellularity, but no blasts or dysplasia. • What is the next best step? 1) Initiate immunosuppressive therapy 2) Continue eculizumab, but change dosing frequency 3) Add darbopoetin alfa 4) Refer for SCT 5) Add an androgen

• A repeat marrow shows 15% cellularity, but no blasts or dysplasia. • What is the next best step? 1) Initiate immunosuppressive therapy 2) Continue eculizumab, but change dosing frequency 3) Add darbopoetin alfa 4) Refer for SCT 5) Add an androgen

Paroxysmal nocturnal hemoglobinuria • Defect in the glycosylphophatidylinositol anchor on the cell membrane. ▫ Loss of the anchor results in absence of GPI linked proteins. ▫ GPI encoded on the PIG-A gene • CD 55: decay accelerating factor • CD 59: membrane inhibitor of reactive lysis

PNH • Result of CD 59 loss: ▫ Sensitivity of red cells to complement mediated hemolysis • Hemolysis is intravascular ▫ Positive DAT (C 3 d) ▫ Negative indirect Coombs • Other symptoms: ▫ Unprovoked clotting, clots in unusual places ▫ Aplasia ▫ Myelodysplasia

PNH • Historically, diagnosed with the Ham test or sucrose lysis test • Now, flow cytometry of RBC or leukocytes evaluating for absence of CD 59 and CD 55 • PNH clones can be found in AA ▫ Need to consider the clinical scenario

PNH • 28% survival at 25 years • Median survival 14 years • Rates: ▫ pancytopenia 15% ▫ myelodysplasia 5% ▫ thrombosis 28% Hillman NEJM 1995: Socie Lancet 1996
: �Binds C 5 complement and inhibits")
PNH • Therapeutic considerations: ▫ Eculizumab (TRIUMPH Trial): �Binds C 5 complement and inhibits terminal complement activation ▫ Results in no need for transfusions ▫ Decreased hemolysis defined by decreased baseline LDH Hillmen NEJM 2006

PNH • Therapeutic considerations: ▫ SHEPERD Study �Similar results as TRIUMPH � 96% of patients free of thrombosis over one year �Decreased need for transfusions ▫ € 300, 000 per year Brodsky Blood 2008

PNH • Housekeeping: ▫ Iron replacement ▫ Folic acid ▫ Prophylactic anti-coagulation: �Retrospective data suggests it is indicated • All therapy is supportive and does not impact the natural history of the underlying disorder. ▫ Unless transplanted
 Ineffective")
Myelodyplastic Syndromes • MDS is a group of diseases characterized by: ▫ 1) Ineffective red cell production ▫ 2) Risk of transformation to leukemia ▫ 3) Disorder arises from transformed hematopoietic stem cell ▫ 4) Lineage decrease resulting in one or more cytopenias • Approximately 10, 000 cases diagnosed per year Aul Br J Hema 1992

Myelodysplastic Syndromes • Median age at diagnosis is greater than 65 with a male predominance. • Associated with: ▫ ▫ ▫ Benzene Trisomy 21 Fanconi Anemia PNH TERT/TERC mutations

MDS- The CBC • Most all patients with MDS will have anemia. ▫ Classically macrocytic • Only 5% of patients with MDS will present with cytopenias WITHOUT anemia. • Thrombocytosis can be present. ▫ 5 q- syndrome ▫ 3 q 21 q 26 syndrome • Thrombocytopenia without anemia or other cytopenias ▫ Think del(20 q) Koeffler 1980

MDS- The Bone Marrow • Characteristic cytogenetic features: ▫ -7 or del 7 q ▫ -5 or del 5 q ▫ del 13 q ▫ del 11 q ▫ del 12 p ▫ del 9 q ▫ t(11; 16) ▫ t(2; 11) • No matter what the blast count: ▫ t(8; 21) ▫ inv 16 ▫ t(15; 17) • This is leukemia and needs to be treated as such

MDS- What else could it be • Idiopathic cytopenia of undetermined significance: ▫ An isolated cytopenia with minimal dysplasia and no cytogenetic abnormalities. � 10% go onto acute leukemia • AML Schroder Ann Onc 2010

MDS- What else could it be • MDS/MPN- A disorder in which dysplasia and proliferation are present. ▫ BCR/Abl negative CML: ‘atypical CML’ �Often characterized by dysplasia in neutrophils ▫ CMML: Overproduction of monocytes �Anemia, thrombocyopenia, splenomegaly �Does not, by definition, have BCR/Abl, PDFR alpha or beta rearrangements

MDS- What else could it be • Aplastic Anemia: ▫ Most patients with MDS have normo/ hypercellular marrow. �Hypocellular marrows with normal cytogenetics: think AA ▫ MDS with hypocellularity: think therapy related MDS • Myelofibrosis: ▫ Marrow fibrosis is common in MDS, sometimes appraching that of PMF ▫ Hyperfibotic MDS: usually no splenomegaly ▫ PMF: 50% will have JAK 2 V 617 F mutation

MDS- What else could it be • HIV▫ Can be hypercellular, dysplastic, fibrotic ▫ Improves long periods of time with good HIV control

MDS Classification • World Health Organization Classification: ▫ Refractory cytopenia with unilineage dysplasia <5% ▫ RA with ringed sideroblasts <5% ▫ Refractory cytopenias with multilineage dysplasia 70% ▫ Refractory anemia with excess blasts 25% ▫ 5 q- 5% ▫ MDS, NOS 5%

MDS Therapy • Most patients are palliative intent • Therapy is indicated if: ▫ Symptomatic anemia ▫ Symptomatic thrombocytopenia ▫ Infection complications from neutropenia

MDS Therapy • Options: ▫ Hypomethylating agents ▫ SCT ▫ Best Supportive Care ▫ Usually limited role for growth factors
: ▫ rare form")
A word on the D-L Antibody • Paroxysmal cold hemoglobinuria (PCH): ▫ rare form of autoimmune hemolytic anemia. ▫ The autoantibody that causes this syndrome is called the Donath Landsteiner antibody. • A biphasic cold hemolysin: ▫ Binds to red blood cells at cold temperatures and causes complement mediated hemolysis after warming to body temperature. ▫ The autoantibody often has specificity for the P blood group antigen. • The test: ▫ Drawing two tubes of blood: � One is incubated at 37 o C for one hour. � The other tube incubated in an ice bath for 30 minutes and then transferred to a 37 o C water bath for an additional 30 minutes. ▫ Both tubes are centrifuged: � If the serum of the tube incubated in the cold is hemoglobin-tinged and the serum of the tube that remained at 37 o C is clear, the patient has a Donath Landsteiner antibody.
- Slides: 68