Bone Marrow Failure Syndromes Brian Boulmay MD Bone

Bone Marrow Failure Syndromes Brian Boulmay, MD

Bone Marrow Failure • Ineffective marrow-poeisis is the final endpoint of many diseases. ▫ Congenital ▫ Acquired �Genetic �Environmental or iatrogenic causes • Congenital marrow failure can present at any age. ▫ Usually in childhood. ▫ Some exceptions • Marrow failure often presents with single lineage declines

Bone Marrow Failure • Early onset of marrow disorders can give a clue to genetic v. acquired syndromes • Dysmorphic body findings ▫ Some may present later in life: �dyskeratosis congenita • Genetic testing will be confirmatory

Inherited Syndromes • Inheritance patterns of inherited forms of marrow failure are variable: ▫ X linked ▫ Autosomal recessive ▫ Autosomal dominant • Not every inherited syndrome has a clear genetic etiology

Inherited Syndromes • Many of the inherited syndromes have an increased risk of acute leukemia and solid tumors. • Penetrance of a disorder may vary within a family.

Marrow morphology • Examination of marrow morphology in a patient with single or multi-lineage cytopenias can be very non-specific. • Often just see decreased marrow elements • Clinical correlates and genetic/molecular analysis is key.

Inherited/Constitutional Causes • • Shwachman- Diamond Syndrome Diamond- Blackfan Anemia Dyskeratosis congenita Fanconi Anemia • Down’s Syndrome • Familial HLH

Shwachman-Diamond Syndrome • Autosomal recessive. • Mutation of SBDS gene on Chr 7. ▫ The SBDS protein is involved with mitotic spindle stabilization ▫ Causes a proliferative defect • Short, pancreatic exocrine deficiency, neutropenia. ▫ May have severe anemia, thrombocytopenia ▫ Longterm increased risk of MDS/AML • Androgens, G-CSF are temporizing measure • SCT will correct the hematologic abnormality

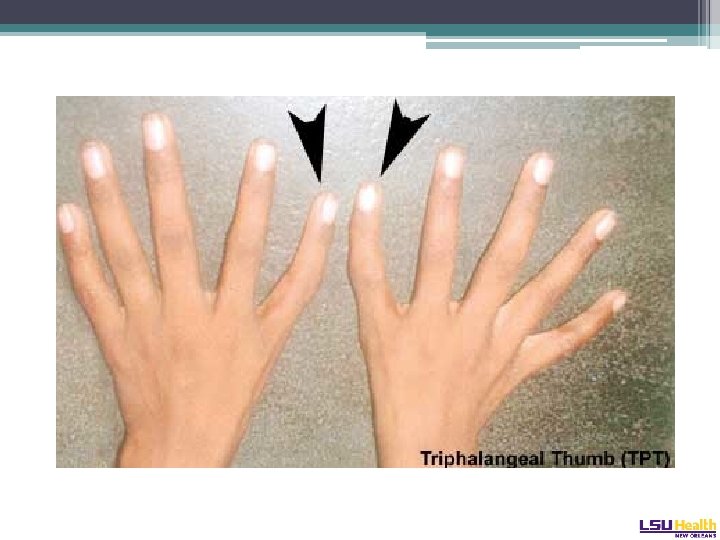
Diamond-Blackfan Syndrome • Autosomal dominant ▫ Pure red cell aplasia • Mutation of the RPS 19 gene on Chr 19 ▫ Plurality of patients, 25% ▫ Involved in ribosome assembly • Considerable variation in presentation: ▫ Hydrops fetalis presentation in adulthood • Physical phenotype: “tow-colored hair, snub nose, wide set eyes, intelligent expression” ▫ Web neck, short stature, triphalangeal thumb Cathie Arch Dis Chil 1950

Diamond-Blackfan Syndrome • Marrow and peripheral blood triad: ▫ ▫ Anemia Reticulocytopenia Absence of red cell precursors in the marrow (Multilineage hypoplasia may develop in long term survivors) • Erythrocyte adenosine deaminase (ADA) levels high • Glucocorticosteroids are the classic therapy ▫ Not effective in all. Adverse effects in children. • SCT the definitive therapy.

Dyskeratosis congenita • X-linked, autosomal dominant or autosomal recessive. • Very rare • Mutations involve genes of telomere maintenance or the telomere complex* ▫ DKC 1 - X linked ▫ TERC- Dominant form ▫ TERT

Dyskeratosis congenita • Physical findings. The mucocutaneous triad: ▫ Leukoplakia ▫ Nail dystrophy ▫ Reticulated skin hyperpigmentation • Can be subtle and only appear later in life.

Dermopath. com, accessed 1/2014

Dyskeratosis congenita • Typically starts with anemia, with high MCV and Hgb F and thrombocytopenia • Progresses to pancytopenia • Median age at presentation is 16 years old • Diagnosis made by flow-FISH of telomere length or quant PCR of telomere DNA or leukocytes
 • Autosomal recessive inheritance •")
Fanconi Anemia • Very rare (1: 1, 000 births) • Autosomal recessive inheritance • Due to mutation in the FANC gene ▫ Protein products of this gene family protect against DNA cross linking. • Most patients present around 8 years old. ▫ Can present in adulthood.

Fanconi Anemia • 60% of patients have a physical exam finding ▫ ▫ Absent radius, absent thumb Short Microcephaly, hypertelorism Skin pigment abnormalities (café au lait spots) • Confirmatory testing: lymphocyte chromosomal breakage on diepoxybutane exposure. ▫ Differentiates from aplastic anemia

Fanconi. co. za, accessed 12/2013

Fanconi Anemia • Therapy: ▫ Androgenic steroids ▫ SCT • Risks: ▫ Increased risk for solid tumors, MDS and AML

Acquired single lineage marrow failure • • • Parvovirus B 19 Thymoma associated pure red cell aplasia Iron Deficiency Anemia of chronic inflammation Nutritional deficiencies ▫ B 12 ▫ Folate • Levamisole tainted cocaine
 • Paroxysmal nocturnal hemoglobinuria • Myelodysplastic")
Acquired multilineage marrow syndromes • Aplastic anemia (autoimmune) • Paroxysmal nocturnal hemoglobinuria • Myelodysplastic syndrome • HIV B 12 def. • Hepatitis C Pregnancy • Bone marrow replacement SLE ▫ Cancer ▫ Myelofibrosis

Aplastic anemia • Frequency 1: 1, 000 • Occurs at any age • Progressive pancytopenia ▫ Often presents with severe pancytopenia • Needs to be differentiated from FA or DC, even in adults. ▫ Consider PNH in differential. • Rule out other marrow injuries ▫ HIV ▫ Hepatitis ▫ Drugs/radiation � Sulfas, seizure meds, HIV meds ▫ ETOH

Aplastic Anemia Cellularity <25% of normal Maturation usually normal Remnant cellularity usually lymphs, plasma cells Consider hypocellular MDS or PNH

Aplastic anemia • On marrow: ▫ No fibrosis ▫ No malignant cells ▫ Hemapoietic precursors do not appear dysplastic • Cytogenetics should be normal

Aplastic Anemia • A population of CD 55 - and CD 59 - cells may be found in patients with AA ▫ Maybe as high as 23% of lymphocytes • Does not mean patient has PNH ▫ Don’t treat as PNH • Predicts response to ATG/Cy. A Sugimori Blood 2006

Aplastic Anemia • Classification: ▫ Moderate aplastic anemia �Bone marrow cellularity <30 percent �Absence of severe pancytopenia �Depression of at least two of three blood elements below normal ▫ Severe aplastic anemia �A bone marrow biopsy showing <25 percent of normal cellularity, or �A bone marrow biopsy showing <50 percent normal �fewer than 30 percent of the cells are hematopoietic and at least two of the following are present: absolute reticulocyte count <40, 000/microliter; absolute neutrophil count (ANC) <500/microliter; or platelet count <20, 000/microliter. ▫ Very severe aplastic anemia �Criteria for severe aplastic anemia are met and the ANC is <200

Aplastic Anemia • Therapy: ▫ Combination of ATG /MP/ Cy. A results in higher response rates than with ATG/ MP alone � 70% v 41% at four months ▫ Lifetime relapse rates ultimately the same in both arms. Frickhofen Blood 2003

Aplastic Anemia

Aplastic Anemia

Aplastic Anemia • Horse ATG versus rabbit ATG: �Primary study endpoint was hematologic response at 6 months: � 68% versus 38% � 3 year OS favored horse ATG � 96% versus 76% Scheinberg NEJM 2011

Aplastic Anemia

Aplastic Anemia • Cy. A should be stopped at 6 months • 2 year taper after the usual 6 months can delay relapse, but does not appear to prevent it. Young Abs 2406 ASH 2011

Aplastic Anemia • Therapy guidelines: ▫ <20 with an HLA matched donor: transplant �Cat 2 C rec for MUD. ▫ 20 - 50 with an HLA matched donor: transplant ▫ >50: immunosuppression

Aplastic Anemia • Role of mimetics ▫ G-CSF and erythropoietin have no effect in AA ▫ Eltrombopag (ELT) is a TPO mimetic: �Triggers the mpl surface receptor. �Approved for use in ITP ▫ 24 patients treated with ELT with refractory AA � 40% response rate ▫ Mpl mutation now implicated in some familial AA Walne Hematologica 2011; Olnes NEJM 2012

Paroxysmal nocturnal hemoglobinuria • Defect in the glycosylphophatidylinositol anchor on the cell membrane. ▫ Loss of the anchor results in absence of GPI linked proteins. ▫ GPI encoded on the PIG-A gene • CD 55: decay accelerating factor • CD 59: membrane inhibitor of reactive lysis

PNH • Result of CD 59 loss: ▫ Sensitivity of red cells to complement mediated hemolysis • Hemolysis is intravascular ▫ Positive DAT (C 3 d) ▫ Negative indirect Coombs • Other symptoms: ▫ Unprovoked clotting, clots in unusual places ▫ Aplasia ▫ Myelodysplasia

PNH • Historically, diagnosed with the Ham test or sucrose lysis test • Now, flow cytometry of RBC or leukocytes evaluating for absence of CD 59 and CD 55 • PNH clones can be found in AA ▫ Need to consider the clinical scenario

PNH • 28% survival at 25 years • Median survival 14 years • Rates: ▫ pancytopenia 15% ▫ myelodysplasia 5% ▫ thrombosis 28% Hillman NEJM 1995: Socie Lancet 1996
: �Binds C 5 complement and inhibits")
PNH • Therapeutic considerations: ▫ Eculizumab (TRIUMPH Trial): �Binds C 5 complement and inhibits terminal complement activation ▫ Results in no need for transfusions ▫ Decreased hemolysis defined by decreased baseline LDH Hillmen NEJM 2006

PNH • Therapeutic considerations: ▫ SHEPERD Study �Similar results as TRIUMPH � 96% of patients free of thrombosis over one year �Decreased need for transfusions ▫ € 300, 000 per year Brodsky Blood 2008

PNH • Housekeeping: ▫ Iron replacement ▫ Folic acid ▫ Prophylactic anti-coagulation: �Retrospective data suggests it is indicated • All therapy is supportive and does not impact the natural history of the underlying disorder. ▫ Unless transplanted
 Ineffective")
Myelodyplastic Syndromes • MDS is a group of diseases characterized by: ▫ 1) Ineffective red cell production ▫ 2) Risk of transformation to leukemia ▫ 3) Disorder arises from transformed hematopoietic stem cell ▫ 4) Lineage decrease resulting in one or more cytopenias • Approximately 10, 000 cases diagnosed per year Aul Br J Hema 1992

Myelodysplastic Syndromes • Median age at diagnosis is greater than 65 with a male predominance. • Associated with: ▫ ▫ ▫ Benzene Trisomy 21 Fanconi Anemia PNH TERT/TERC mutations

MDS- The CBC • Most all patients with MDS will have anemia. ▫ Classically macrocytic • Only 5% of patients with MDS will present with cytopenias WITHOUT anemia. • Thrombocytosis can be present. ▫ 5 q- syndrome ▫ 3 q 21 q 26 syndrome • Thrombocytopenia without anemia or other cytopenias ▫ Think del(20 q) Koeffler 1980

MDS- The Bone Marrow • Characteristic cytogenetic features: ▫ -7 or del 7 q ▫ -5 or del 5 q ▫ del 13 q ▫ del 11 q ▫ del 12 p ▫ del 9 q ▫ t(11; 16) ▫ t(2; 11) • No matter what the blast count: ▫ t(8; 21) ▫ inv 16 ▫ t(15; 17) • This is leukemia and needs to be treated as such

MDS- What else could it be • Idiopathic cytopenia of undetermined significance: ▫ An isolated cytopenia with minimal dysplasia and no cytogenetic abnormalities. � 10% go onto acute leukemia • AML Schroder Ann Onc 2010

MDS- What else could it be • MDS/MPN- A disorder in which dysplasia and proliferation are present. ▫ BCR/Abl negative CML: ‘atypical CML’ �Often characterized by dysplasia in neutrophils ▫ CMML: Overproduction of monocytes �Anemia, thrombocyopenia, splenomegaly �Does not, by definition, have BCR/Abl, PDFR alpha or beta rearrangements

MDS- What else could it be • Aplastic Anemia: ▫ Most patients with MDS have normo/ hypercellular marrow. �Hypocellular marrows with normal cytogenetics: think AA ▫ MDS with hypocellularity: think therapy related MDS • Myelofibrosis: ▫ Marrow fibrosis is common in MDS, sometimes appraching that of PMF ▫ Hyperfibotic MDS: usually no splenomegaly ▫ PMF: 50% will have JAK 2 V 617 F mutation

MDS- What else could it be • HIV▫ Can be hypercellular, dysplastic, fibrotic ▫ Improves long periods of time with good HIV control

MDS Classification • World Health Organization Classification: ▫ Refractory cytopenia with unilineage dysplasia <5% ▫ RA with ringed sideroblasts <5% ▫ Refractory cytopenias with multilineage dysplasia 70% ▫ Refractory anemia with excess blasts 25% ▫ 5 q- 5% ▫ MDS, NOS 5%

MDS Therapy • Most patients are palliative intent • Therapy is indicated if: ▫ Symptomatic anemia ▫ Symptomatic thrombocytopenia ▫ Infection complications from neutropenia
: ▫ Percentage of Bone Marrow Blasts � <5 percent")
MDS Therapy • IPSS (old): ▫ Percentage of Bone Marrow Blasts � <5 percent (0 points) � 5 to 10 percent (0. 5 points) � 11 to 20 percent (1. 5 points) � 21 to 30 percent (2. 0 points) ▫ Karyotype � Normal, Y-, 5 q-, 20 q- (0 points) � Abnormal chromosome 7 or 3 or more abnormalities (1. 0 points) � All other cytogenic abnormalities (0. 5 points) ▫ Cytopenias (defined as hemoglobin <10, ANC<1800, platelet count <100 K) � No cytopenia or cytopenia of 1 cell type (0 points) � Cytopenia of 2 or 3 cell types (0. 5 points)

MDS Therapy- The IPSS Score • 0 Points: Low (Median Survival of 5. 7 yrs) • 0. 5 -1 Points: Intermediate 1 (Median Survival of 3. 5 yrs. ) • 1. 5 -2. 0 Points: Intermediate 2 (Median Survival of 1. 2 yrs. ) • 2. 5 -3. 5 Points: High (Median Survival of 0. 4 yrs. )

MDS Therapy • Options: ▫ Hypomethylating agents ▫ SCT ▫ Best Supportive Care ▫ Usually limited role for growth factors
: ▫ rare form")
A word on the D-L Antibody • Paroxysmal cold hemoglobinuria (PCH): ▫ rare form of autoimmune hemolytic anemia. ▫ The autoantibody that causes this syndrome is called the Donath Landsteiner antibody. • A biphasic cold hemolysin: ▫ Binds to red blood cells at cold temperatures and causes complement mediated hemolysis after warming to body temperature. ▫ The autoantibody often has specificity for the P blood group antigen. • The test: ▫ Drawing two tubes of blood: � One is incubated at 37 o C for one hour. � The other tube incubated in an ice bath for 30 minutes and then transferred to a 37 o C water bath for an additional 30 minutes. ▫ Both tubes are centrifuged: � If the serum of the tube incubated in the cold is hemoglobin-tinged and the serum of the tube that remained at 37 o C is clear, the patient has a Donath Landsteiner antibody.
- Slides: 55