Bleeding disorders 2482020 Bleeding Disorders Abnormal coagulation cascade

Bleeding disorders 24/8/2020

Bleeding Disorders • Abnormal coagulation cascade Hemophilia, Vitamin K deficiency • Abnormal platelets Bernard-Soulier Glanzmann’s Thrombasthenia ITP, TTP , HUS , • Mixed Disorders Von Willebrand Disease, DIC

Type of Bleeding • Abnormal platelets Mucosal bleeding, skin bleeding, petechiae • Abnormal coagulation factors - Joint bleeding, deep tissue bleeding

Labs tests : 1 - Bleeding Time • Test of platelet function 2 - PTT 3 - PT 4 - Thrombin Time
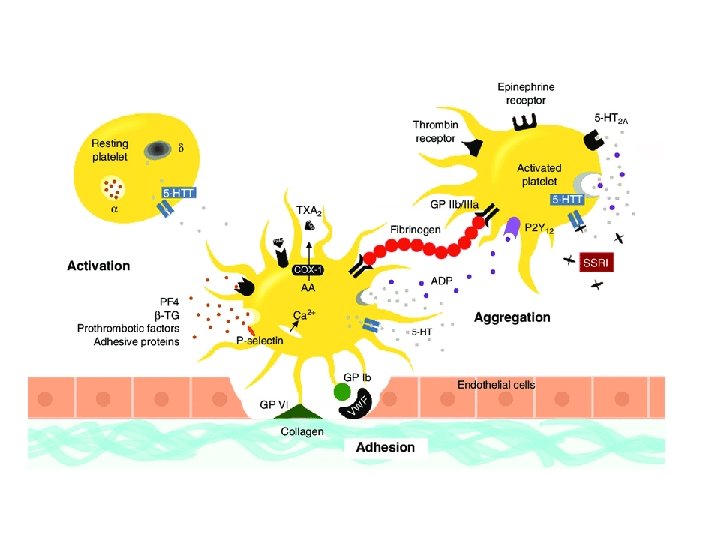

Inherited Platelet Disorders Glanzmann’s Thrombasthenia : -Autosomal recessive disorder -deficiency of GPIIb/IIIa receptors -present with bleeding symptoms in childhood. -Blood smear: Isolated platelets (no clumping) -Absent platelet aggregation in response to stimuli -platelet aggregometry -Bernard-Soulier Syndrome -Autosomal recessive disorder -present with bleeding symptoms in childhood. -deficiency of GPIb plt receptors : - Platelets cannot bind v. WF -Also results in large platelets Key lab findings: Prolonged bleeding time -Thrombocytopenia -- Large platelets on blood smear - Wiskott-Aldrich Syndrome : -plt and Immunodeficiency syndrome of infants - X linked disorder of WAS gene (WAS protein) * Necessary for T-cell cytoskeleton maintenance • Immune dysfunction ↓ platelets Eczema

Giant Platelets Can be seen in association with thrombocytopenia
 *immune-mediated destruction of platelets")
acquired platelet disorder 1 - ITP (Idiopathic thrombocytopenic purpura ) *immune-mediated destruction of platelets and possibly inhibition of platelet release from the megakaryocyte caused by anti-GPIIB/IIIA antibodies : Consumption in splenic macrophages Splenomegaly Thrombocytopenia In children, it is usually an acute disease, most commonly following an infection, and with a self-limited course. In adults, it is a more chronic disease, although in some adults, spontaneous remission occurs,

Diagnosis of exclusion • Rule out other causes of bone marrow suppression characterized by : mucocutaneous bleeding and a low, often very low, platelet count, with an otherwise normal peripheral blood cells and smear. - Laboratory testing for antibodies (serologic testing) is usually not helpful due to the low sensitivity and specificity of the current tests -Large plts on perepheral smear • Treatment: Initial treatment in patients without significant bleeding symptoms, severe thrombocytopenia (<5000/μL) or signs of impending bleeding (such as retinal hemorrhage or large oral mucosal hemorrhages) can be instituted as an outpatient usingle agents. - Steroids - IVIG (blocks Fc receptors in macrophages) - Splenectomy

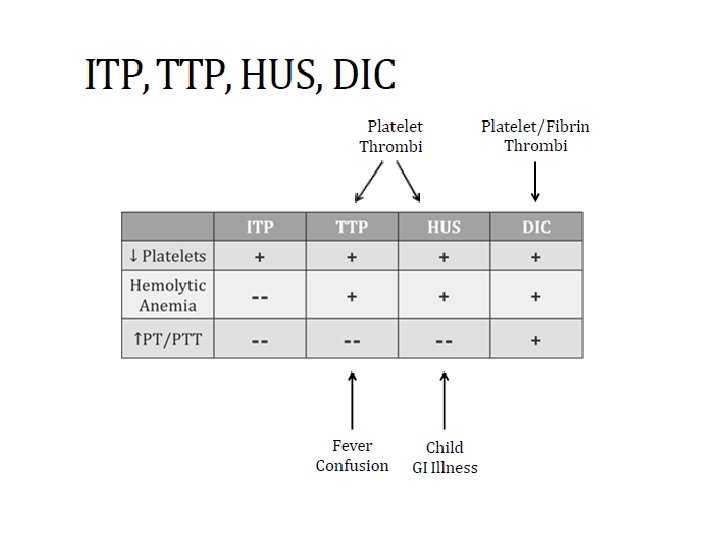
2 - TTP Thrombotic thrombocytopenic purpura • Disorder of small vessel thrombus formation • Consumes platelets ==> thrombocytopenia • ↓ activity of v. WF cleaving protease ADAMTS 13

Cause • Severe ADAMTS 13 deficiency , Usually <10% normal activity • Usual cause: acquired autoantibody to ADAMTS 13 - Result: v. WF multimers in areas of high shear stress , - Obstruction small vessels Microangiopathic hemolytic anemia • Hemolytic anemia (↑LDH, ↓ haptoglobin) • Caused by shearing of RBCs as they pass through thrombi in small vessels • Blood smear: • Seen in: TTP HUS DIC schistocytes

Clinical symptoms : • Fever • Neurological symptoms : Headache, confusion, seizures • Renal failure • Petechiae and bleeding Non-specific Lab tests: • Hemolytic anemia • Thrombocytopenia • Schistocytes on blood smear • PT/PTT should be normal • Contrast with DIC • May see elevated d-dimer Treatment • Plasma exchange: removes antibodies • Platelet counts monitored to determine efficacy

3 -HUS Hemolytic Uremic Syndrome • Many similarities with TTP • Also caused by platelet-rich thrombi in small vessels • MAHA, thrombocytopenia, acute kidney injury (Renal thrombi => kidney injury ) • Usually no fever or CNS symptoms • Commonly seen in children , follow GI infection E. Coli( Shiga-like toxin causes microthrombi ) Treatment of HUS is primarily supportive. In HUS associated with diarrhea, many (~40%) children require at least some period of support with dialysis Plasma infusion or plasma exchange has not been shown to alter the overall course. ADAMTS 13 levels are generally reported to be normal in HUS In patients with a. HUS, eculizumab, a humanized monoclonal antibody against C 5 that blocks terminal complement, has efficacy in resolution of HUS and improving or preserving renal function

DIC Disseminated Intravascular Coagulation • Widespread activation of clotting cascade leading to Diffuse thrombi (platelets/fibrin) => ischemia • Consumption of clotting factors and platelets • Destruction of red blood cells=> anemia • Elevated PT/PTT/Thrombin time - Consumption of factors • Low platelets - Consumption of platelets • Low fibrinogen (consumption) • Microangiopathic hemolytic anemia • Low RBC (anemia) • Schistocytes on blood smear

Occurs secondary to another process : • Obstetrical emergencies : ( Amniotic fluid contains tissue factor) : abruptio placenta , major haemorrhagic bleeding , can develop DIC. • Sepsis : 1 - Endotoxin –> activates coagulation cascade , 2 - Cytokines • Cancer: (well-described hypercoagulable state) Excess coagulation: DIC -Leukemia : acute promyelocytic leukemia (APML) • Rattlesnake bites : Thrombin-like glycoproteins within venom • Treatment: underlying disorder • Fresh frozen plasma: replace clotting factors • RBCs, platelets • Cryoprecipitate (for low fibrinogen)

Von Willebrand Disease Most common inherited bleeding disorder : Affects up to 1 percent of population • Gene mutations => ↓ level or function of v. WF • Most cases autosomal dominant (males=females) Clinical symptoms : Usually mild, non-life-threatening bleeding • Easy bruising • Skin bleeding • Prolonged bleeding from mucosal surfaces -Severe nosebleeds - Menorrhagia Diagnosis • Normal platelet count • Normal PT • Increased PTT (depending on severity) Usually no joint/deep tissue bleeding • Increased bleeding time

Diagnosis • Ristocetin cofactor activity assay Treatment • v. WF concentrate • Desmopressin : Analogue of vasopressin (ADH) with no pressor activity , Increases v. WF and factor VIII levels , Releases v. WF from endothelial cells • Aminocaproic acid : Antifibrinolytic drug , Inhibits plasminogen activation => plasmin = Less breakdown of formed clots

Heyde’s Syndrome?

Hemophilias • X-linked recessive diseases • Gene mutations: Run in families; also occur de novo( 30%) • Hemophilia A: Deficiency of factor VIII • Hemophilia B: Deficiency of factor IX ( Also called Christmas disease )
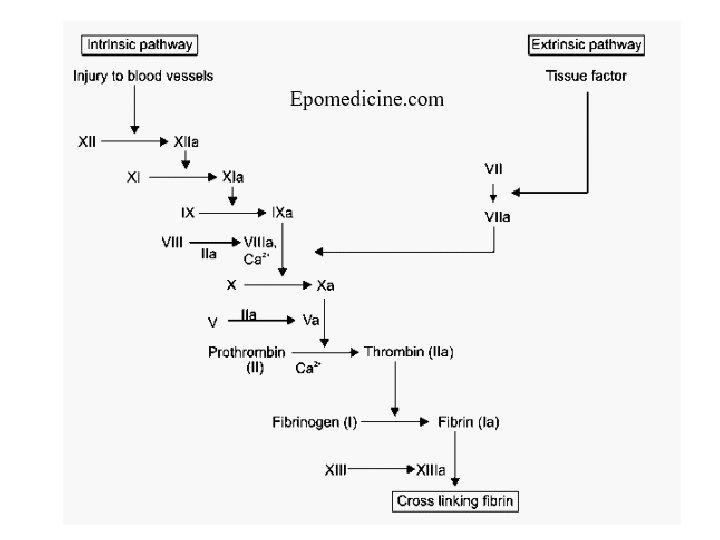

• Present with spontaneous or easy bruising • Recurrent joint bleeds is common presentation • Screening: PTT will be prolonged Factors VIII, IX both part of intrinsic pathway • PT, bleeding time, platelet count all normal

Treatment 1 - Replacement factor VIII and IX 2 -NONTRANSFUSION THERAPY IN HEMOPHILIA * Desmopressin (d. DAVP) • Used in mild hemophilia A • Antifibrinolytic Drugs : Aminocaproic acid

Coagulation Factor Inhibitors • Antibodies , Inhibit activity or increase clearance of clotting factor • Inhibitors of factor VIII most common • Often occur in association with: - Malignancy - Post-partum - Autoimmune disorders Can be treated with prednisone Can present similar to hemophilia • Deficient activity of VIII= bleeding , Prolonged PTT • Mixing study will differentiate from hemophilia A

Vitamin K Deficiency • Results in bleeding • Deficiency of vitamin K-dependent factors : II, VII, IX, X • Key lab findings: Elevated PT/INR , Can see elevated PTT (less sensitive) • Normal bleeding time *Dietary deficiency rare : (: GI bacteria produce sufficient quantities) • Common causes: -Warfarin - Antibiotics (deplete GI bacteria) - Newborns (sterile GI tract) - Malabsorption (Vitamin K is fat soluble)

Thank you
- Slides: 26