Bleeding and coagulation disorders Dr Mrinalini Kotru Prof

Bleeding and coagulation disorders Dr Mrinalini Kotru Prof Pathology, UCMS & GTBH

Homeostasis Heamostasis
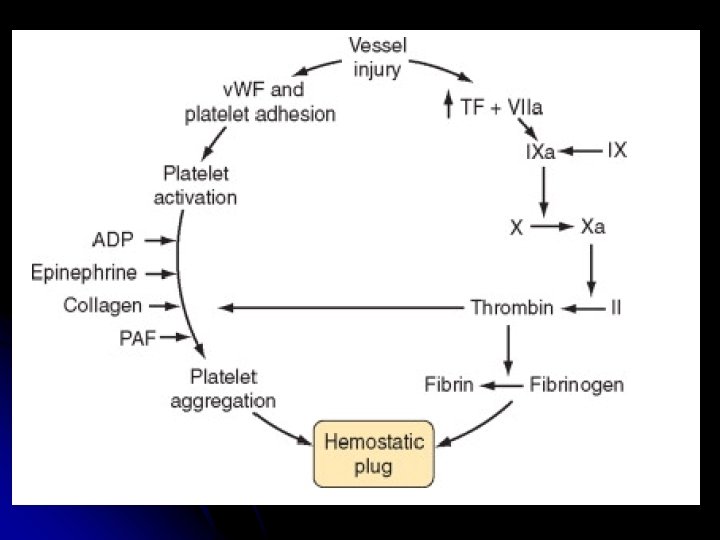

Components of haemostasis l Blood vessel wall l l Coagulation factors Platelets Primary haemostasis Secondary Haemostasis

Disorders of hemostasis l Disorders of primary hemostasis Vessel wall abnormalities l Platelet disorders l l Qualitative l Quantitive l Disorders of secondary hemostasis l Hereditary l Hemophilia l VWD l Acquired

Clinical Presentation
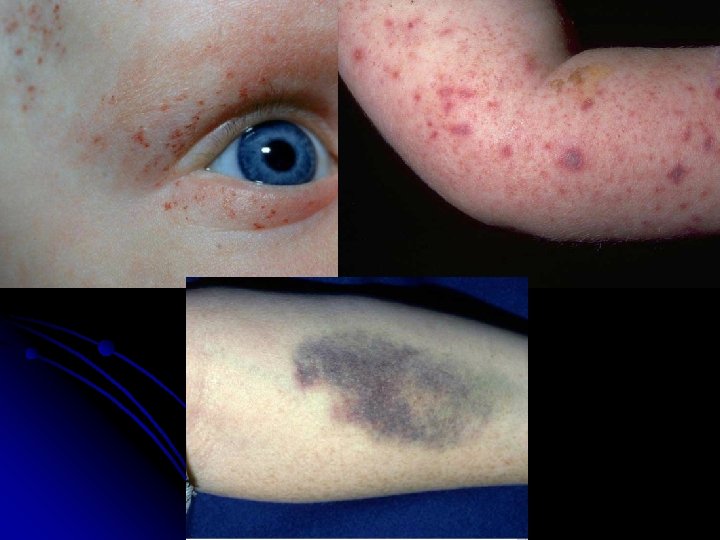

6 yr old boy presenting with multiple ecchymotic patches and B/L knee swelling

Approach to diagnosis l Detailed clinical history l Type of bleeding l Disorder of primary vs. secondary hemostasis l Hereditary l vs Acquired Laboratory evaluation of increased bleeding tendency l Screening coagulogram

Clinical features

Screening Coagulogram Platelet count l PT l APTT l l Additional l Bleeding time l TT l S. Fibrinogen 11

Analysis Bleeding time l Platelet count l PT l APTT l S. Fibrinogen l Must for Screening for increased bleeding tendency

Platelet count l Automated cell counters l Different methods for analysis l Impedance l Optical – better l Flourescence l Flowcytometry l Many times falsely low l Confirm on peripheral smear

Prothrombin time l Measures the time for plasma to clot in presence of Tissue Factor and Calcium l 2, 7, 10 (Vit K dependent factors) l Extrinsic & intrinsic pathway

l Normal -11 -16 sec Thromboplasti n
")
l Causes l Oral anticoagulants l Liver disease l Vitamin K def (obstructive jaundice) l DIC l Factor 7 (isolated prolongation) l Factor 2, 5, 10

Activated Partial Thromboplastin Time l Measures time taken by plasma to clot by activating the intrinsic pathway. calcium l Kaolin l l -activate the contactdependent Factor XII l Cephalin l l substitutes for platelet phospholipids Normal 26 -40 secs.

Activated Partial Thromboplastin Time l Tests for intrinsic pathway l Deficiencies of all factors except factor 7 l Inhibitors l Heparin monitoring l DIC l Liver disease l Massive transfusion

Thrombin time Time taken for the plasma to form clot on addition of thrombin l Normal 15 -19 sec /within 2 sec of control l Causes of prolongation l l Hypofibrinogenemia l Dysfibrinogenemia l FDPs l Heparin (RVV normal) l Hypo/paraproteinemia

l Shortened TT l Faulty l collection Marked elevation l Heparin contamination l Marked increased concentration of S. Fibrinogen

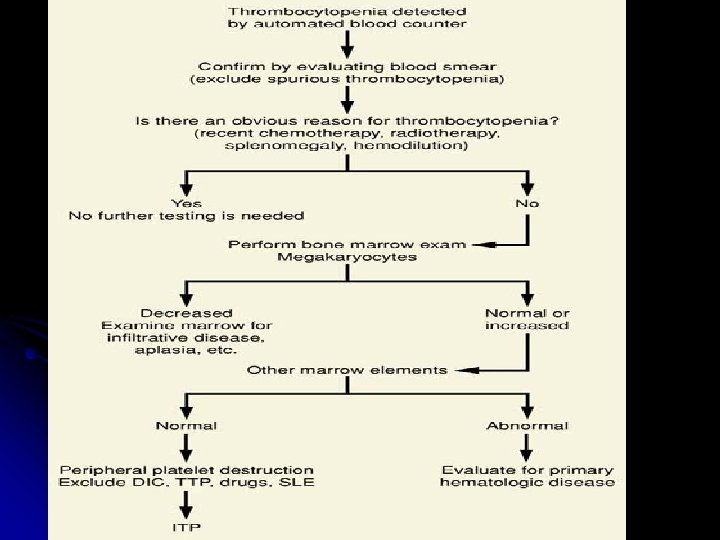
What to do with an abnormal result? Further testing

Interpretation Workup

How to collect specimens? l Anticoagulant l 3. 2 % Trisodium Citrate l Vacutainer l Tube l made in the laboratory Sealed with para film

Diseases to be discussed Thrombocytopenia l Hemophilia l VWD l DIC l

Thrombocytopenia

Thrombocytopenia Normal count: 150 -450 x 109/L l Platelets remain in the circulation for 8 -10 days l l Thrombocytopenia – when platelet count <150 x 109/L

The pathophysiology of thrombocytopenia


Thrombocytopenia Associated with HIV Infection Thrombocytopenia is common. Various causes of thrombocytopenia include: Ø Reduced platelet production. Ø Accelerated platelet destruction Ø splenic sequestration, and Ø Rarely due to Platelet consumption associated with thrombotic thrombocytopenic purpura (TTP). Medications, Concurrent infections such as hepatitis C, and Hematologic malignancies

Nutritional Deficiencies and Alcohol-Induced Thrombocytopenia l Associated with megaloblastic anemia resulting from vitamin B 12 deficiency and folic acid. l Alcoholics l l liver cirrhosis with relative TPO deficiency, congestive splenomegaly, and/or from folic acid deficiency. some patients(alcoholic), primarily from direct marrow suppression.

THROMBOCYTOPENIA RESULTING FROM ACCELERATED PLATELET DESTRUCTION l Immune l l l Autoimmune alloimmune Non-Immune l Thrombotic microangiopathies

Autoimmune thrombocytopenic purpura Premature destruction of Platelets autoantibody or immune complex deposition on their membranes. Site of destruction: - Reticulo-endothelial system Spleen Liver bone marrow (less common).

Causes
 l Thrombocytopenia in which apparent exogenous etiologic factors are lacking")
Immune Thrombocytopenic Purpura (ITP) l Thrombocytopenia in which apparent exogenous etiologic factors are lacking and in which diseases known to be associated with secondary thrombocytopenia have been excluded.

Incidence l Annual incidence l l l 5. 5 per 100, 000 persons when cutoff platelet count <100 x 109/L 3. 2 per 100, 000 using a cutoff platelet count <50 x 109/L. The annual incidence of ITP is about 3 to 8 cases per 100, 000 children with a peak in the 2 to 5 year age group.

Acute ITP l l Isolated thrombocytopenia Resolves spontaneously most often in <6 months is termed “Acute” The incidence peaks in the winter and spring, following the incidence of viral infections. Approximately 7 to 28% of children with acute ITP develop the chronic variety
")
Chronic ITP l l Lasting >6 months and requiring therapy to improve thrombocytopenia. (WINTROBE’S) According to INDIAN HEMOPHILIA AND THROMBOSIS CENTRE: - l l l If ITP has persisted >3 months If it has not responded to a splenectomy and the platelet count has been <50, 000 cells/mm 3. In the pediatric setting, however, the designation for chronic ITP is used only with duration of disease of ≥ 6 months.


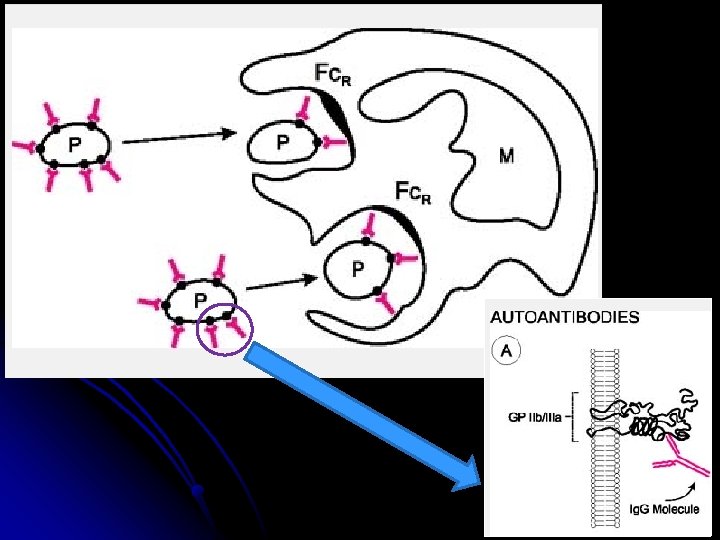
Pathophysiology l caused by platelet-specific autoantibodies l l l bind to autologous platelets rapidly cleared from the circulation by the mononuclear phagocyte system via macrophage FcγRIIA receptors predominantly in the spleen, liver and bone marrow. Activation of components of complement on the platelet surface are also demonstrated

Common presenting symptoms in patients with ITP <1 %
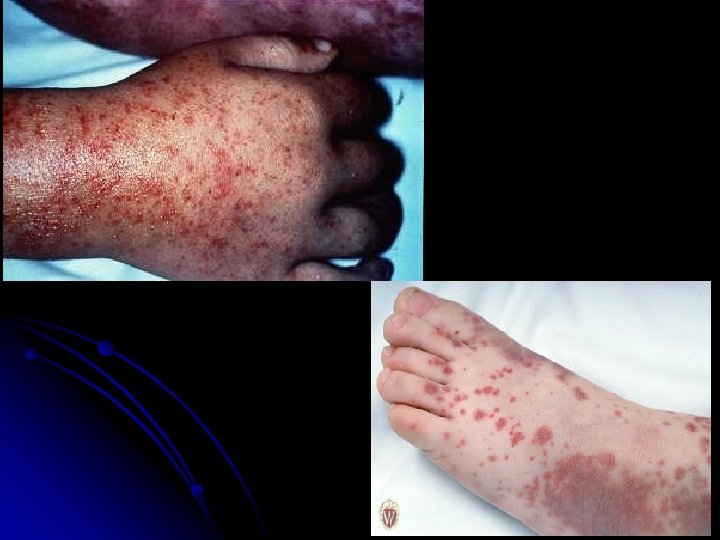
 are common. The")
Blood: Laboratory findings Abnormalities in platelet size and morphology (platelet anisocytosis) are common. The platelets often are abnormally large (3 to 4 μm in diameter) and reveal more than normal variation in size and shape.

Diagnosis l Anemia, if present, is proportional to the extent of blood loss and is usually normocytic l “Evans syndrome”: Thrombocytopenia and Coomb’s positive hemolytic anemia and have no other known underlying etiology. l Prolonged bleeding time. l The results of tests of blood coagulation (PT, a. PTT) are normal.

Bone marrow Megakaryocytes usually are ↑in size ↑ or normal in number “Smooth” forms with single nuclei, scanty cytoplasm and relatively few granules are common It results markedly accelerated platelet production and the presence of many young forms. Normoblastic hyperplasia may develop as a result of blood loss. The leukocytes are essentially normal with the exception of occasional eosinophilia


BMA often not indicated. It is indicated in 1. 2. Atypical clinical symptoms: Presence of malaise, lymphadenopathy, hepatosplenomegaly or other cytopenias. Age: 1. 2. 3. age >60 years to rule out MDS Pediatric age to rule out leukemia Refractory ITP: If patients do not respond to therapy, to exclude other hematological disorders.

THROMBOTIC MICOANGIPATHIES
. Microangiopathic hemolytic anemia 2). Thrombocytopenia")
q Thrombotic microangiopathy refers to a combination of 1). Microangiopathic hemolytic anemia 2). Thrombocytopenia 3). Microvascular thrombosis, regardless of cause or specific tissue involvement
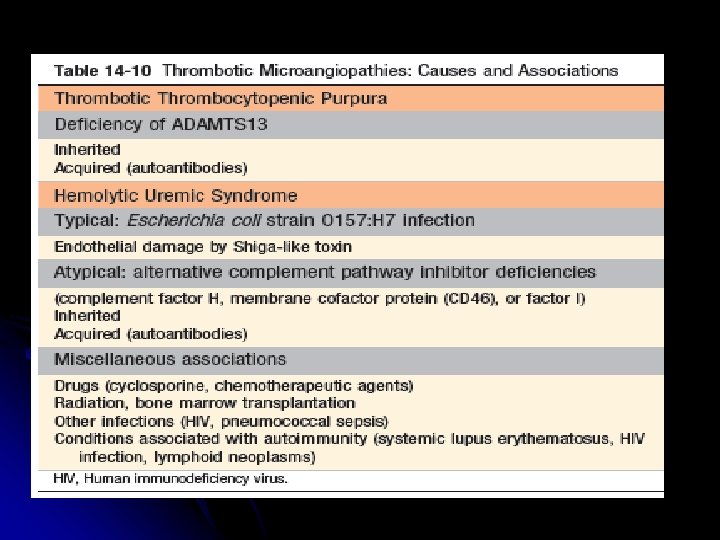
. Hemolytic uremic syndrome a) b) Diarrhea positive (Infectious, Shiga toxin-associated) Sporadic / Epidemic")
3). Hemolytic uremic syndrome a) b) Diarrhea positive (Infectious, Shiga toxin-associated) Sporadic / Epidemic Diarrhea negative: -Inherited complement regulatory protein deficiencies (factor H, membrane cofactor protein, factor I) 4). Secondary thrombotic microangiopathy

TTP "Pentad" of signs and symptoms: l Thrombocytopenia l Microangiopathic hemolytic anemia; l Renal failure l Neurologic abnormalities l Fever

Classification l v v v Three distinct types of TTP are recognized: Congenital TTP – severe deficiency of ADAMTS 13 Idiopathic TTP – autoantibodies to ADAMSTS 13 Non- idiopathic TTP- associated with malignancy, drugs, pregnancy etc

PATHOPHYSIOLOGY l Unregulated v. WF dependent platelet thrombosis appear to be the mechanism of TTP. l Pathologic hallmark: Microvascular occlusion of terminal arteries and capillaries by platelet and v. WF rich thrombus (having NO THROMBIN)
-dependent platelet thrombosis by ADAMTS 13")
Regulation of von Willebrand factor (VWF)-dependent platelet thrombosis by ADAMTS 13

PATHOGENESIS OF TTP Absence or decreased level of ADAMTS 13 â No timely cleavage of ULv. WF multimers â Uncleaved ULv. WF anchors to the endothelial cells via Pselectin molecules â Passing platelets adhere via GPIb receptor â Induce adhesion and subsequent aggregation of platelets â Large, potentially occlusive platelet thrombi
")
LABORATORY FEATURES l l l Anemia (1/3 have Hb less than 6 g/dl ) Elevated reticulocyte count Thrombocytopenia (half of patient have platelets below 20, 000/µL ) Increased serum lactate dehydrogenase level Decreased serum haptoglobin level

Peripheral blood film

Coomb’s test is almost always negative. Normal plasma fibrinogen, prothrombin time and activated partial thromboplastin time ADAMTS 13 related tests ADAMTS 13 activity levels ADAMTS 13 antigen levels Anti-ADAMTS 13 autoantibodies

At the end of this lecture you should know the following-

l To describe the normal coagulation pathway l Differences between bleeding due to platelet disorders and coagulation factor deficiency. l l Define Bleeding time (BT) and Clotting time (CT) and describe methods of estimation l List the causes of prolonged BT and CT
 and Activated partial thromboplastin time (APTT)")
l. Describe the principle of Prothombin time (PT) and Activated partial thromboplastin time (APTT) tests. l. Enumerate the causes of increased PT and PTT l. List the causes of thrombocytopenia
. l Differences between")
l Etiopathogenesis, clinical features and lab diagnosis of Immune thrombocytopenia (ITP). l Differences between acute and chronic ITP. l Differences between bleeding due to platelet disorders and coagulation factor deficiency List the diseases causing non- immune thrombocytopenia. l

 Etipathogenesis, clinical features and lab diagnosis of Thrombotic thrombocytopenic purpura (TTP) and")
12) Etipathogenesis, clinical features and lab diagnosis of Thrombotic thrombocytopenic purpura (TTP) and Hemolytic uremic syndrome (HUS) 13) Short note on Von Willebrand disease (VWD)

- Slides: 67