Bioqumica heptica El metabolismo del glucgeno Sntesis y

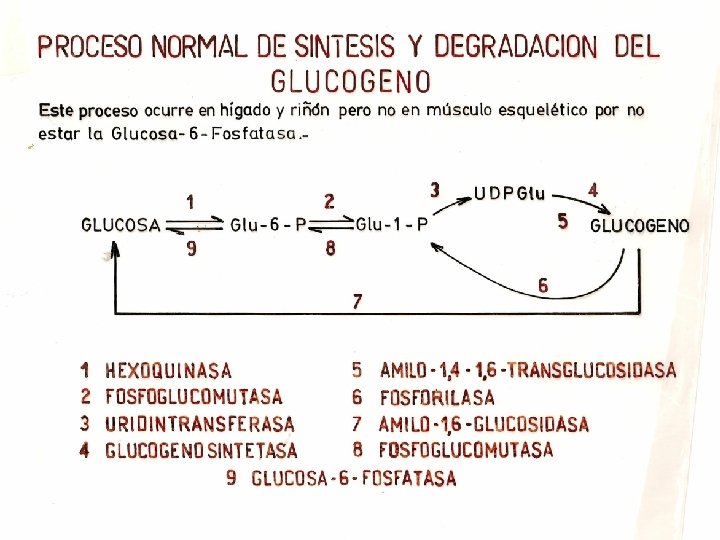
Bioquímica hepática El metabolismo del glucógeno. Síntesis y degradación

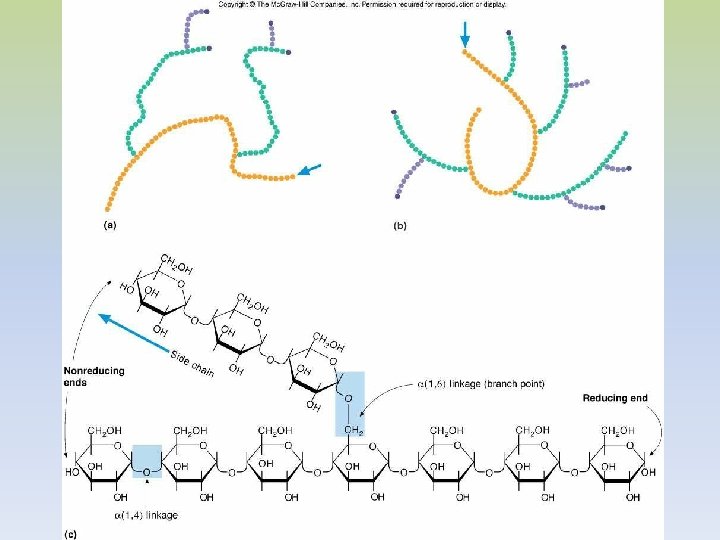
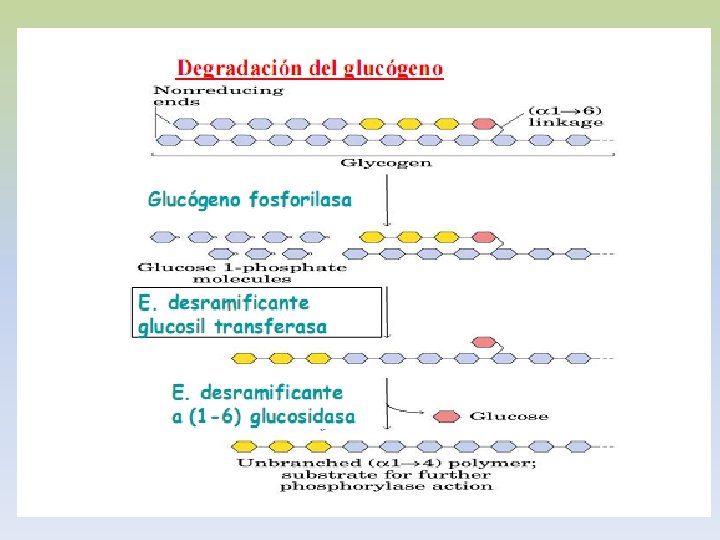
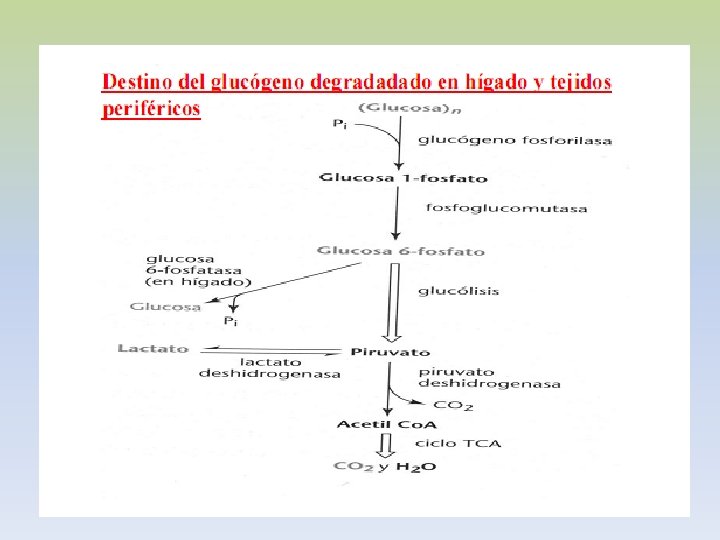
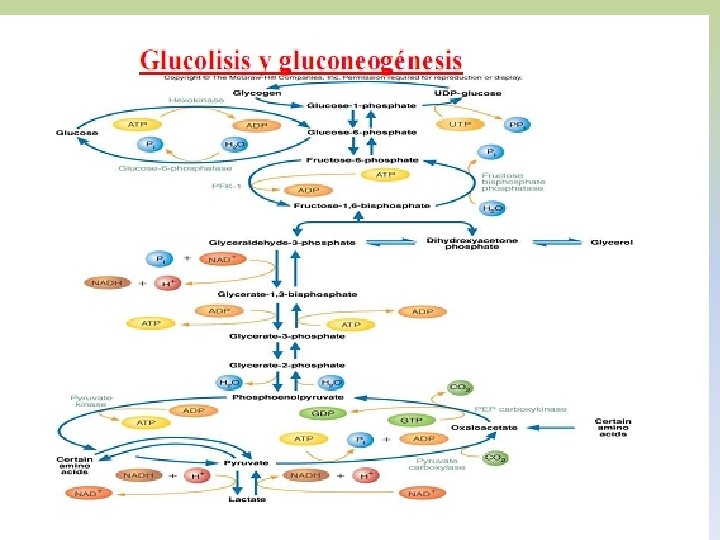
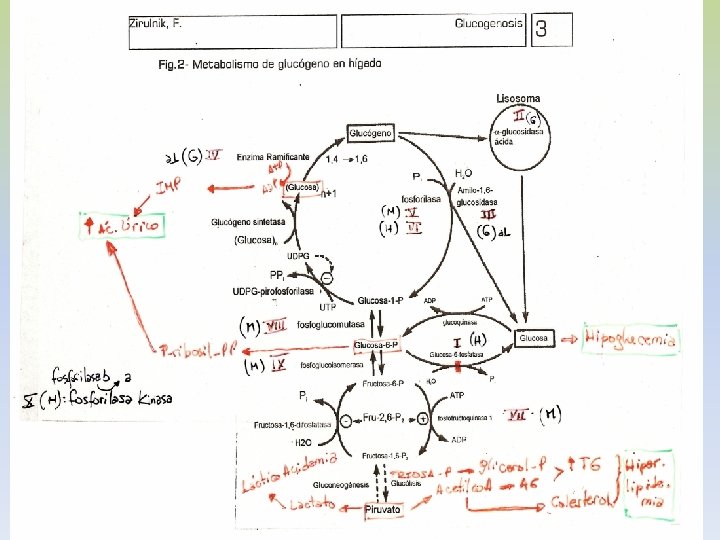
-EL GLUCÓGENO ES UN POLÍMERO MACROMOLECULAR, ALTAMENTE RAMIFICADO. -SOLO GLUCOSA ES SU CONSTITUYENTE BÁSICO. -PRESENTA UNIONES ALFA 1, 4 Y ALFA 1, 6. -EXISTE GLUCÓGENO HEPÁTICO Y MUSCULAR. -EN HÍGADO EL GLUCÓGENO SE ENCUENTRA EN EL CITOPLASMA, EN FORMA DE GRÁNULOS, EN LOS QUE TAMBIEN SE ENCUENTRAN LAS ENZIMAS DE SU METABOLISMO. -LA CANTIDAD DE GLUCÓGENO HEPÁTICO, CONTRIBUYE A REGULAR EL NIVEL DE GLUCOSA EN LA SANGRE, YA QUE PROVEE UN DEPÓSITO DE GLUCOSA QUE ES DE FÁCIL ACCESO, EN CASO DE NECESIDAD.
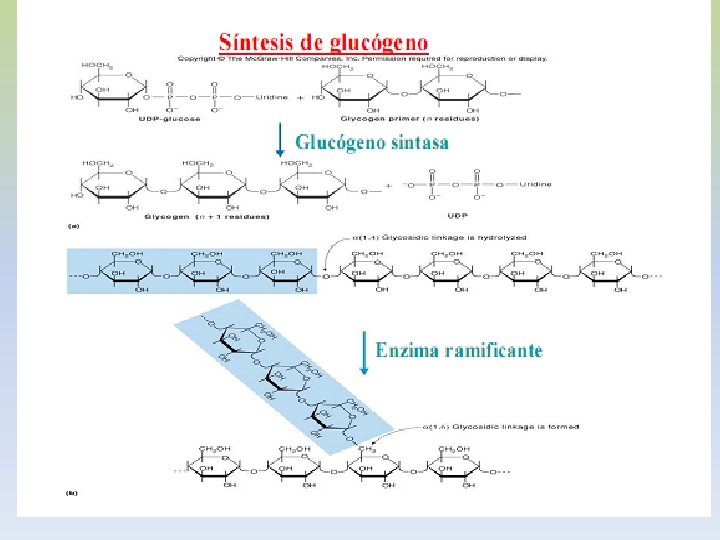
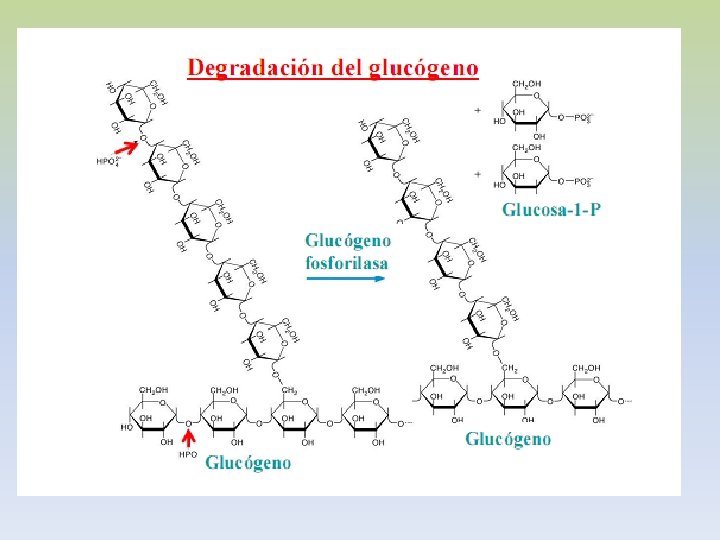

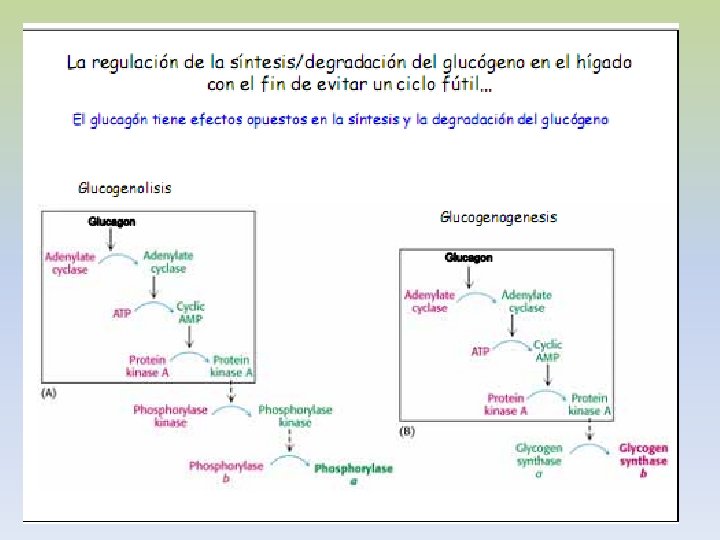

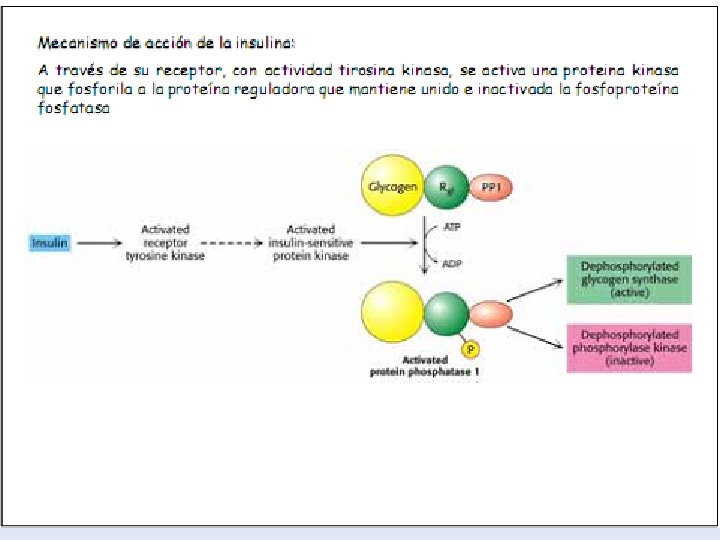
-La insulina que se libera del páncreas en respuesta a una elevación de la glucemia, desencadena una cascada de activación de la fosfoproteína fosfatasa. -Esta fosfatasa cataliza la hidrólisis de los grupos fosfato de todos los enzimas implicados en el metabolismo del glucógeno (fosforilasa, fosforilasa quinasa, sintetasa). -El resultado es la inactivación de la glucogenolisis y la activación de la glucogenogénesis. -La síntesis eficaz de glucógeno tendrá lugar en la medida haya UDP-glucosa disponible. -La insulina antagoniza los efectos del glucagón, porque aumenta el transporte de glucosa hacia los tejidos.

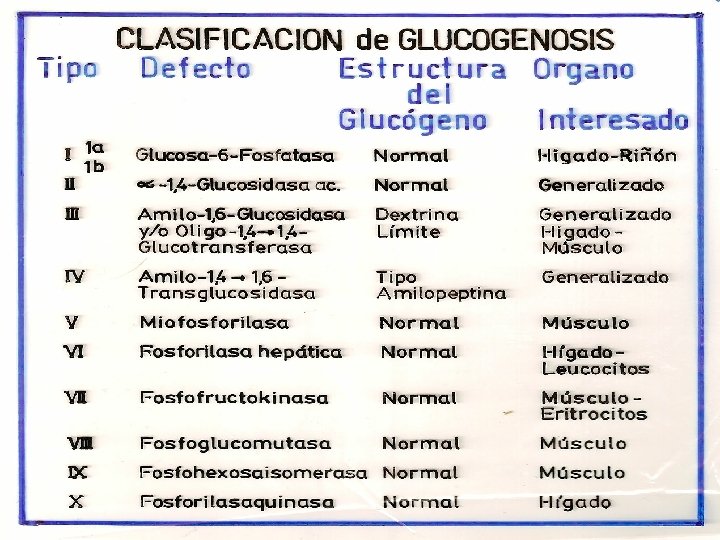
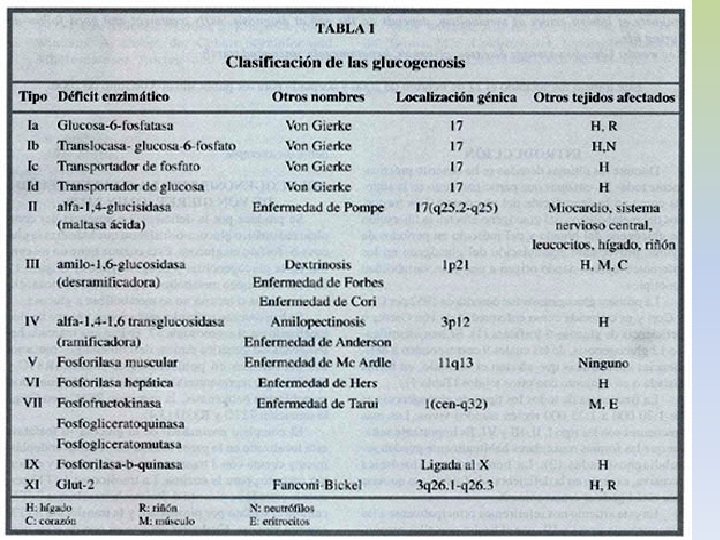
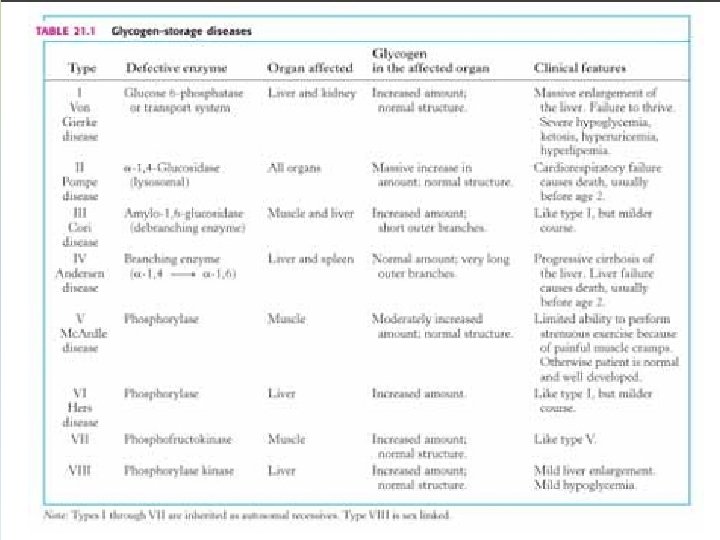
GLUCOGENOSIS -TRASTORNOS EN EL METABOLISMO DEL GLUCÓGENO POR AUSENCIA DE ENZIMAS QUE PARTICIPAN EN SU BIOSÍNTESIS Y DEGRADACIÓN -SE TRADUCE EN UNA ANOMALÍA CUANTITIATIVA O CUALITATIVA DEL GLUCÓGENO DEPOSITADO. -SEGÚN EL ÓRGANO AFECTADO LAS GLUCOGENSOSIS SE CLASIFICAN EN: I) HEPÁTICA O HEPATORRENAL: I, III, IV, VI, X HEPATOMEGALIA, HIPOGLUCEMIIA, HIPERTG, HIPERCOLESTEROLEMIA, LACTATO Y ÁCIDO ÚRICO. MUERTE INFANCIA II) CARDÍACA O GENERALIZADA: II INFILTRACIÓN MASIVA CON GLUCÓGENO, ESPECIALMENTE MIOCARDIO, FUNCIÓN MUSCULAR MUERTE FALLA CARDÍACA III) MIOPÁTICA: V, VIII, IX - CALAMBRES, CANSANCIO, ATROFIA MUSCULAR

GLUCOGENOSIS TIPO I: ENFERMEDAD DE VON GIERKE • Incapacidad de liberar glucosa al torrente sanguíneo a partir de Glucosa-6 -P • Déficit de G-6 -Pasa - AR • Manifestación hepática: hepatomegalia, riñón agrandado • Hipoglucemia severa durante el ayuno inhibe la liberación de insulina • Aumento de la glucolisis hepática, sin liberación de glu al torrente sanguíneo • Acidemia severa hipoglucemia estimula la liberación de catecolaminas y facilita la degradación de glucógeno muscular a ácido láctico. • Infiltración grasa por liberación de lípidos como fuente de energía • Menor síntesis de proteínas crecimiento retardado. • Grave
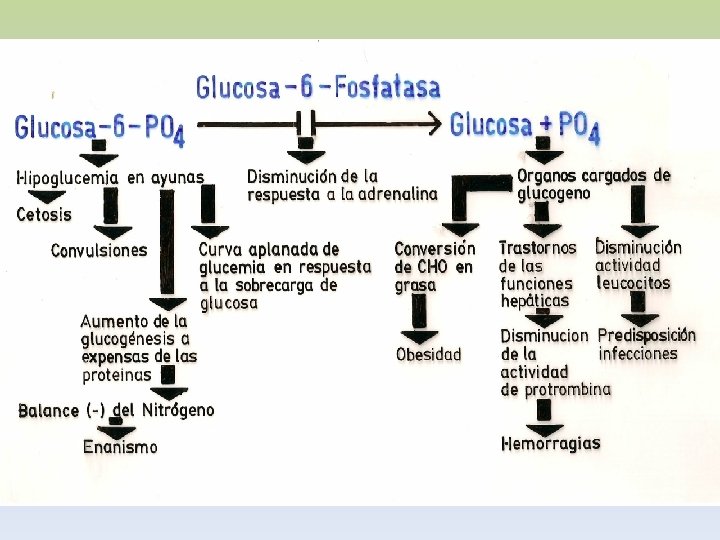

TRATAMIENTO: - INGESTIÓN FRECUENTE DE ALIMENTOS Y UN RÉGIMEN RICO EN PROTEÍNAS EVITARÍA O DISMINUIRÍA EL DEPÓSITO DE GLUCÓGENO. ACTH O CORTISONA PARA REGULAR LOS EPISODIOS DE HIPOGLUCEMIA. -PARA EVITAR LA ACIDOSIS ADM LACTATO DE SODIO. -DIAGNÓSTICO: - SE DEMUESTRA LA AUSENCIA DE LA ENZIMA EN TEJIDO HEPÁTICO POR BIOPSIA O EXÁMEN POST-MORTEM. -PRUEBAS DE TOLERANCIA: A LA GALACTOSA, FRUCTOSA, GLUCAGÓN Y EPINEFRINA NO SE OBSERVA AUMENTO DE LA GLUCEMIA

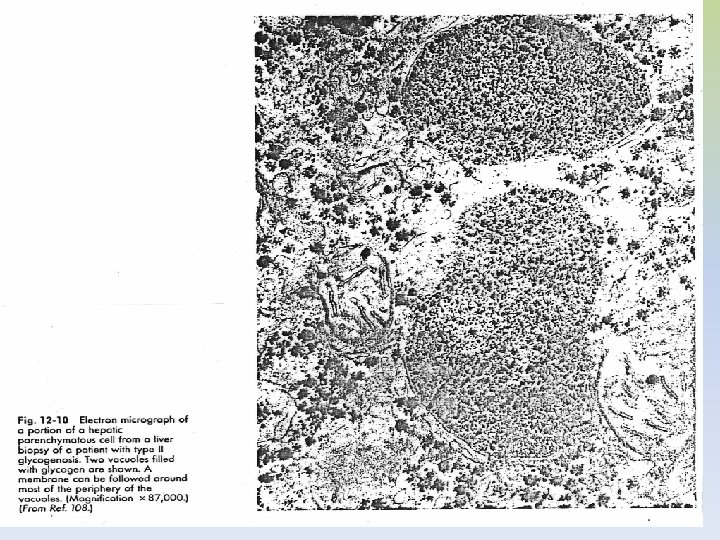
GLUCOGENOSIS TIPO II: ENFERMEDAD DE POMPE • Ausencia de alfa 1 -4 glucosidasa lisosomal – AR • Se manifiesta en la primera infancia Síntomas clínicos: vómitos, anorexia, retardo de crecimiento, debilidad muscular, cianosis, disnea. • Las alteraciones metabólicas no son muy pronunciadas dado que las otras vías metabólicas del glucógeno no están afectadas • Cardiomegalia que puede producir la muerte a edad temprana por fallo cardíaco Observaciones físicas: apariencia de imbecilidad, hipotonía muscular, hipertrofia cardíaca, alteraciones neurológicas, no se observa hepato-esplenomegalia. • Acumulación de rosetas de glucógeno no degradadas en el interior de los lisosomas • No hay tratamiento

Diagnóstico: -La glucemia y las pruebas de tolerancia dan todas normales -Cuadro hematológico: normal, la tinción de leucocitos para identificar glucógeno revela acumulación del mismo. -Alteraciones cardiográficas -No se observa: hipoglucemia, acidosis y cetosis, lípidos plasmáticos normales. -Se observa acumulación de glucógeno en corazón y músculo.
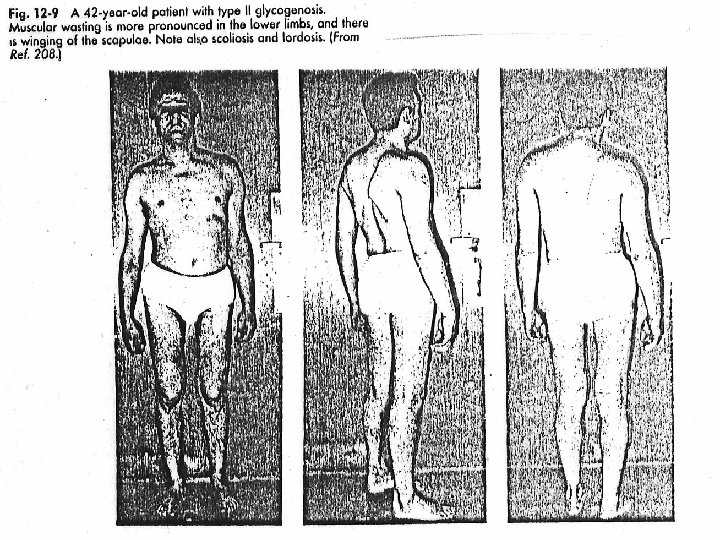

GLUCOGENOSISTIPO III: • Ausencia del enzima desramificante amil-1, 6 -glucosidasa - AR • El glucógeno solo se puede degradar parcialmente por la glucógeno fosforilasa, no más allá de la dextrina límite. • Manifestaciones clínicas: • Afectados hígado y músculo • Más moderadas que en la enfermedad de von Gierke. • Hipoglucemia menos severa • Acumulación de glucógeno hepático, que tiene cadenas externas cortas. TRATAMIENTO: - Dieta rica en proteínas la transaminación y conversión de aminoácidos en intermediarios glucolíticos puede corregir el déficit primario.

DIAGNÓSTICO: -Si damos Gal o Fru aumenta la glucemia porque está la Glu-6 -Pasa y puede pasar a Glu ( a la tipo I) -DOBLE PRUEBA DEL GLUCAGÓN: -La administración de glucagón no aumenta la glucemia en la tipo I. En la tipo III -A) La prueba se hace luego de una hora de ayuno glucógeno se encuentra ramificado si es tipo III aumenta la glucemia -B) después de 14 hs de ayuno, se reanuda la adm de glucagón no aumenta la glucemia porque por la primera prueba, el glucógeno llegó a los puntos de ramificación. -DETERMINACION ENZIMÁTICA: SUSTRATO ARTIFICIAL PENTASACÁRIDO (4 GLU- ALFA 1, 4 Y UNA GLU TERMINAL CON UNIÓN ALFA 1, 6

GLUCOGENOSIS TIPO IV: DEFICIT DE LA ENZIMA RAMIFICANTE O AMILOPECTINOSIS -NO SE SABE SI ES AR O LIG A X -AUSENTE LA ENZIMA AMILO 1, 4 - 1, 6 – TRANSGLICOSIDASA -SÍNTOMAS CLINICOS: AL AÑO DE EDAD CIRROSIS HEPÁTICA, HÍGADO GRANDE, NODULAR, ESPLENOMEGALIA. -PRUEBAS FUNCIONALES HEPÁTICAS ANORMALES -CARACTERÍSTICAS ESTRUCTURALES DEL GLUCÓGENO AISLADO DE TEJIDOS: -Similar a la amilopectina, es decir con menor número de puntos ramificados (comparado con el glucógeno normal) -Menos soluble que el glucógeno normal -Cirrosis por reacción contra un cuerpo extraño glucógeno precipitado. -No se observa acumulación de glucógeno en tejidos y GR. -Grave el paciente muere antes de los 5 años. -TRATAMIENTO: SINTOMÁTICO Y PALIATIVO

DIAGNÓSTICO: - Si se mide la absorción de amilopectina con I 2 se observa un pico máximo a 550 nm -El glucógeno de los enfermos + I 2 presenta un pico a 530 nm. - El glucógeno normal + I 2 470 nm la estructura del glucógeno del enfermo es similar a la amilopectina -La enzima tb se encuentra ausente en leucocitos se hacen ensayos “in vitro”, para demostrar la ausencia de esta enzima -Las respuestas a la adrenalina, glucagón y sobrecarga a la glu son anormales.
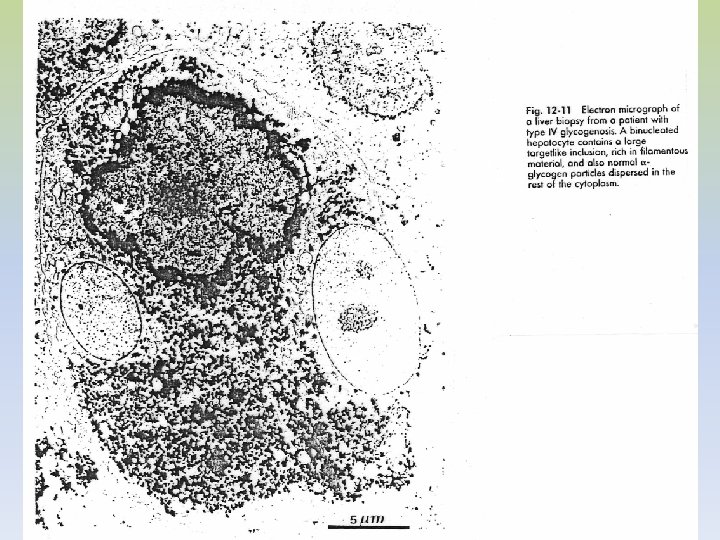

GLUCOGENOSISTIPO V: ENFERMEDAD DE MC ARDLE • AR - Ausencia de fosforilasa muscular • Fosforilasa hepática normal distinto control genético • No se moviliza el glucógeno muscular como consecuencia del ejercicio, su estructura es normal. • En esta enfermedad faltan las fosforilasas a (activa) y b (inactiva) SÍNTOMAS CLÍNICOS: • Incapacidad para el esfuerzo físico, astenia, disfagia. • Los paciente sufren calambres y la imposibilidad de realizar ejercicios mínimamente enérgicos • No se observa el aumento de lactato sérico típico después de un esfuerzo muscular • Daño muscular (distrofia) como consecuencia de un metabolismo energético inadecuado • Aumento de marcadores de lesión muscular (CPK, aldolasa, mioglobina). TRATAMIENTO: -No hay

DIAGNÓSTICO: -Un enfermo no puede hacer flexiones profundas de rodillas durante más de 1 minuto. -Se hace la biopsia muscular y se mide la concentración de glucógeno y actividad de fosforilasa. -PRUEBA DEL EJERCICIO ANÓXICO: -Se insufla el manguito del aparato de presión arterial por encima de la presión sistólica. -A- Se obtiene una muestra de sangre venosa antes del ejercicio. -B- Se abre y se cierra el puño durante un minuto. -C- Se extrae nuevamente sangre -D- Se repite 3 veces -Los niveles sanguíneos de lactato se elevarán aproximadamente el 3 veces en un sujeto normal -En el paciente no se observa lo mismo queda su mano en posición tetánica

Glucogenosis. Tipo VI: • Ausencia de fosforilasa hepática – AR • No se moviliza el glucógeno hepático • La hipoglucemia moderada en ayunas, compensada en parte por gluconeogénesis Lípidos moderadamente aumentados Glucógeno en GR aumentado Äcido úrico aumentado La administración de Gal, aumenta la glucemia DIAGNÓSTICO: -Deter la actividad de fosforilasa en leucitos e hígado -Cuantificar contenido de glucógeno. -La activ de la enzima es normal luego de la adm de glucagón -TRATAMIENTO: -- Dieta rica en proteínas y adm de zinc- glucagón -La fosforilasa hepática es genéticamente diferente a la muscular.

GLUCOGENOSIS TIPO VII: -Deficiencia de la fosfofructoquinasa muscular -Dolores musculares -DIAGNÓSTICO: - El músculo contiene mas del doble de glucógeno que lo normal. -Su estructura es normal -La producción de lactato a partir de glucógeno, Glu- 1 -P y Fru-1 -P es baja. -Su producción desde Fru-1, 6 di. P es normal -La ausencia de la enzima ha sido determinada por reacción inmunológica. GLUCOGENOSIS TIPO VIII: -Deficiencia de fosfoglucomutasa -Hay acumulación de glucógeno en músculo -Hepatomegalia GLUCOGENOSIS IX: -Deficiencia de fosfohexosa isomerasa muscular -Se mide lactato con distintos sustratos usando homogenatos de músculo.

GLUCOGENOSIS TIPO X: -Deficiencia de fosforilasa quinasa de hígado AGLUCOGENOSIS: -Ausencia de glucógeno sintetasa -Bajos niveles de glucógeno -Hipoglucemia, marcada en ayunas -Deficiencia mental secundaria a la hipoglucemia -Alteración en el metabolismo de los lípiods -Diagnóstico: biopsia ausencia de glucógeno y glucógeno sintetasa
- Slides: 37