BIOLOGICAL SYSTEM A Gastrointestinal and Hepatic Systems Regulation

BIOLOGICAL SYSTEM A Gastrointestinal and Hepatic Systems Regulation of Drug Transport, Absorption, Distribution, Excretion and Metabolism Dennis Paul, Ph. D. Department of Pharmacology 504 -568 -4740 rwainf@lsuhsc. edu

Principles of Pharmacology Common processes and mechanisms whereby: • Drugs gain access to the body • Drugs move throughout the body • Drugs produce an effect by altering a physiological process • Drugs are removed from the body

Pharmacokinetics The study of drug movement into, within and out of the body, which includes absorption, distribution, metabolism and elimination

• Pharmacokinetics: • Absorption: Transfer of drug from site of administration to systemic circulation • Distribution: Transfer of drug from systemic circulation to tissues • Metabolism: Alteration of drug to increase excretion from the body • Excretion: Drug movement out of the body
 Enteral – directly")
Routes of drug administration Affects onset and duration of drug 1) Enteral – directly into G. I. tract Oral or rectal administration - safest, cheapest, most convenient - slow onset, sometimes unpredictable response 2) Parenteral – bypasses G. I. tract Usually injection, can be inhalation or topical administration - fast absorption, rapid onset, predictable response - more expensive, more difficult, painful, requires sterile conditions
 ● Advantages -Most common, convenient, painless and inexpensive way to administer")
Oral route (enteral) ● Advantages -Most common, convenient, painless and inexpensive way to administer a variety of drugs e. g. liquid, tablet, coated tablet etc -GI tract a large blood rich absorbent surface ● Disadvantages -First pass metabolism Drug must pass through GI tract and liver before entering circulation and therefore are subject to metabolism meaning higher doses are given orally e. g. morphine -Food and GI motility affects absorption – must comply with instructions e. g. with food or on empty stomach -Can be difficult to predict percentage of active drug that reaches patient
 Usually suppository, cream or enema e. g. aspirin, barbituates Drug mixed")
Rectal route (enteral) Usually suppository, cream or enema e. g. aspirin, barbituates Drug mixed with waxy substance that dissolves in the rectum Advantages -Reduced first pass metabolism, some rectal veins lead into direct circulation bypassing liver -Used in patients unable to take drugs orally e. g. elderly, young, unconscious Disadvantages -Not well liked by patients -Absorption very variable so not reliable method of drug delivery
![[Drug] Drug Profiles Time (h) Physical Properties Structure Lipid solubility Ionization state](http://slidetodoc.com/presentation_image_h/b91c160ecb0510e4ac16c704a05bf745/image-9.jpg "[Drug] Drug Profiles Time (h) Physical Properties Structure Lipid solubility Ionization state")
[Drug] Drug Profiles Time (h) Physical Properties Structure Lipid solubility Ionization state

Objectives I. III. IV. V. Mechanisms of drug transport Drug absorption Drug distribution Drug metabolism Drug excretion

Mechanisms of Drug Transport 1. Passive diffusion a. Passive diffusion of non-electrolytes b. Passive diffusion of electrolytes 2. Filtration 3. Carrier-mediated transport a. Active transport b. Facilitated diffusion 4. Receptor-mediated endocytosis 5. Ion-pair transport Endogenous compounds and drugs

Mechanisms of Drug Transport 1. Passive diffusion – Low molecular weight drugs that are both water and lipid soluble dissolve in membrane and cross to the other side. Primary means by which drugs cross membranes

Mechanisms of Drug Transport • Passive Diffusion • Drugs dissolve and cross the cell membrane following concentration gradient. • Characteristics of drugs that use passive diffusion: ● Small ● Predominantly lipid soluble ● Uncharged ● Small water soluble molecules pass via pores
 Passive diffusion of non-electrolytes 2) Passive")
Mechanisms of Drug Transport 1. Passive diffusion 1) Passive diffusion of non-electrolytes 2) Passive diffusion of electrolytes – Influence of p. H and pka – Weak acids are uncharged in acidic environment – Weak bases are uncharged in basic environment
 Passive diffusion of non-electrolytes Lipid-water partition")
Mechanisms of Drug Transport 1. Passive diffusion 1) Passive diffusion of non-electrolytes Lipid-water partition coefficient (Kp) - the ratio of the concentration of the drug in two immiscible phases: a nonpolar liquid (representing membrane) and an aqueous buffer (representing the plasma). Kp can be measured. Kp = [drug] in lipid phase/[drug] in aqueous phase. If the drug is more soluble in the lipid, Kp is higher. If the drug is more soluble in the aqueous phase, Kp will be lower. The partition coefficient is a measure of the relative affinity of a drug for the lipid and aqueous phases. One can control the Kp by modifying the side groups on the compound. The more C and H on the compound, the more lipid soluble, and thus the higher the Kp. The more O, S and the more water-soluble the compound, and the lower the Kp.

Mechanisms of Drug Transport Passive diffusion of non-electrolytes: The higher the Kp, the more lipid soluble, the faster the rate of transfer across biological membranes
 Passive diffusion of electrolytes p. Ka: the p. H")
Mechanisms of Drug Transport 2) Passive diffusion of electrolytes p. Ka: the p. H at which half of the molecules are in the ionized form and one half are in the unionized form. p. Ka is a characteristic of a drug. Henderson-Hasselbalch equations: For acids: p. H = p. Ka + log [A-]/[HA] For bases: p. H = p. Ka + log [B]/[BH+]
 Passive diffusion of electrolytes HA H+ + A -")
Mechanisms of Drug Transport 2) Passive diffusion of electrolytes HA H+ + A - BH+ H+ + B p. H < p. Ka Predominate forms: HA and BH+ p. H 3 4 p. H > p. Ka p. H = p. Ka 5 HA = ABH+ = B 6 7 Predominate forms: A- and B 8 9 10 11
 Passive diffusion of electrolytes Only the unionized forms of")
Mechanisms of Drug Transport 2) Passive diffusion of electrolytes Only the unionized forms of the drug or the uncharged drug can pass through or across the membranes (or is transferred) by passive diffusion. - Un-ionized form acts like a nonpolar lipid soluble compound and can cross body membranes - Ionized form is less lipid soluble and cannot easily cross body membranes By controlling the p. H of the solution and/or the p. Ka of the drug, you can control the rate at which the drug is transferred

Mechanisms of Drug Transport - Drugs that are weak electrolytes equilibrate into ionized and non-ionized forms in solution - p. H (H+ concentration) at site of administration and the dissociation characteristics (p. Ka) of the drug determine the amount ionized and non-ionized drug
 100 mg")
Mechanisms of Drug Transport ASPIRIN p. Ka = 4. 5 (weak acid) 100 mg orally H+ + A - 1 = [ I ] Stomach p. H = 2 Blood p. H = 7. 4 99 = [ UI ] HA Aspirin is absorbed from stomach (fast action)

Mechanisms of Drug Transport Aspirin accumulation Acidic drug - p. Ka = 4. 5 [ UI ] 1 HA p. H = 2 Body compartment 1 - stomach [I] 0. 01 H+ + A - Membrane Body compartment 2 – blood p. H = 7. 4 HA 1 [ UI ] H+ + A 100 [I]
 100 mg")
Mechanisms of Drug Transport STRYCHNINE p. Ka = 9. 5 (weak base) 100 mg orally H+ + B 99 = [ I ] Stomach p. H = 2 Blood p. H = 7. 4 1 = [ UI ] HB+ Strychnine not absorbed until enters G. I. tract

Mechanisms of Drug Transport 2. Filtration - Passage of molecules through membrane pores or porous structures.

Mechanisms of Drug Transport The rate of filtration a. Driving force: The pressure gradient in both sides. b. The size of the compound relative to the size of the pore. i. Smaller compound – transfer rapidly ii. Larger compound – retained iii. Intermediate compound – barrier Lipid soluble – passive diffusion Water soluble – filtration

Mechanisms of Drug Transport The rate of filtration: In biological systems: Filtration is the transfer of drug across membrane through the pores or through the spaces between cells a. Capillary endothelial membranes b. Renal glomerulus • Most substances (lipid-soluble or not) – cross the capillary wall – very fast • Lipid soluble and unionized – filtration and passive diffusion – at the same time

Mechanisms of Drug Transport • . 3 Carrier-mediated transport A. Active transport - Goes against concentration gradient - Requires energy (ATP) - Mediated by transport carrier proteins - Drug combines with a transport protein in the membrane and the complex can move across the membrane a. Selectivity - not for all drugs b. One-way process – against drug concentration gradient resulting in drug accumulation c. It can be saturated – Drug/receptor ratio – enzyme-catalyzed reactions d. Can be inhibited – ATP inhibitors, structural analogous compounds

Mechanisms of Drug Transport 3. Carrier-mediated transport b. Facilitated diffusion a. b. c. d. e. f. Carrier or receptor-mediated Selective to specific molecules e. g. glucose It can be saturated Does not require ATP Does not go against the concentration gradient Bi-directional – no drug accumulation

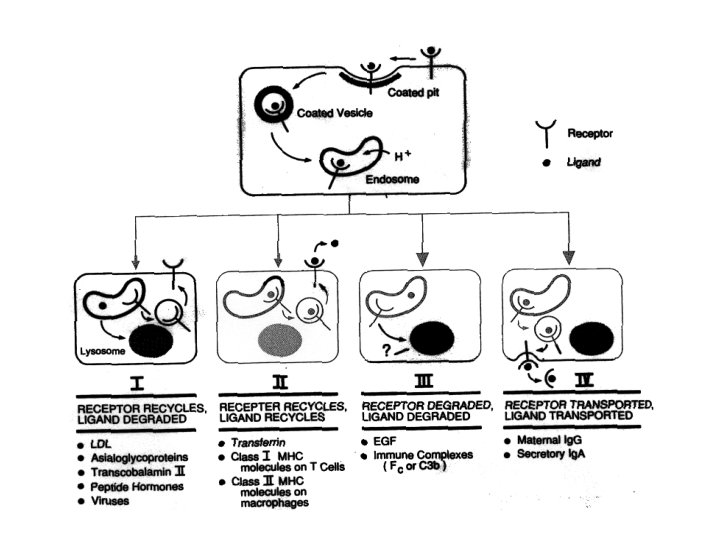
Mechanisms of Drug Transport 4. Receptor-mediated endocytosis - more specific uptake process Drugs (peptide hormones, growth factors, antibodies, et al. ) bind to their receptors on the cell surface in coated pits, and then the ligand receptors are internalized, forming endosomes. Receptor-ligand complex may take four different pathways: a. b. c. d. Receptor recycles, ligand degraded Receptor and ligand recycle Receptor and ligand degraded Receptor and ligand transported

Mechanisms of Drug Transport 5. Ion-pair transport Highly ionized Passive diffusion + + + _ Carrier _ + _ _

Pharmacokinetics: Absorption • Process by which drug molecules are transferred from administration site to systemic circulation • Factors affecting absorption: 1. site of GI absorption 2. modifications • Bioavailibility • Solubility of drug e. g. suspension absorbed more slowly than a solution, solubility = absorbance
 Mouth 2) Stomach")
Drug Absorption 1. Sites of absorption through the GI tract 1) Mouth 2) Stomach 3) Small intestine 4) Large intestine
 Mouth: a. Small amount of surface area but good blood flow")
Drug Absorption 1) Mouth: a. Small amount of surface area but good blood flow – best for potent drugs. b. Transfer by passive diffusion – good for lipid soluble drugs. c. p. H = 6. Weak base drugs have better absorption. Nicotine p. Ka 8. 5 p. H Ionization Absorption Mouth 6 less ionized 4 times faster d. Can bypass first pass effect. GI tract 1 -5 ionized
 Stomach: a. Moderate surface area – more than mouth, less than")
Drug Absorption 2) Stomach: a. Moderate surface area – more than mouth, less than small intestine. b. Good blood supply. c. Drugs absorbed in the stomach will experience first pass effect. d. Transfer by passive diffusion. e. Low p. H (1 -2) – ionization - Drugs that are weak acids will be absorbed better than weak base drugs. f. Ion trapping: Accumulation of weak base drugs in the stomach.
 Small intestine a. The primary site for most drugs. b. Large")
Drug Absorption 3) Small intestine a. The primary site for most drugs. b. Large surface area - Folds, villi and microvilli. c. p. H = 5 -8. d. Passive diffusion. e. Absorption can also take place by active transport, facilitated diffusion, endocytosis and filtration.
 Large intestine a. Not important for drug absorption, if the drug")
Drug Absorption 4) Large intestine a. Not important for drug absorption, if the drug is absorbed effectively in small intestine. b. Can be a site of absorption for incompletely absorbed drugs. c. Less absorption then small intestine – less area and solid nature of contents.
 Drug solubilization")
Drug Absorption 2. Factors that modify absorption in the GI tract 1) Drug solubilization 2) Formulation factors 3) Concentration of drug at the absorption site 4) Blood flow at the absorption site 5) Surface area of absorption small intestine 6) Route of administration 7) Gastric emptying 8) Food 9) Intestinal motility 10) Metabolism of drug by GI tract
 Drug solubilization – breaking drugs into smaller, more absorbable particles Hydrophilic")
Drug Absorption 1) Drug solubilization – breaking drugs into smaller, more absorbable particles Hydrophilic drugs - poorly absorbed - inability to cross the lipid-rich cell membrane. Hydrophobic drugs - poorly absorbed - insoluble in the aqueous body fluids - cannot gain access to the surface of cells. - largely hydrophobic yet have some solubility in aqueous solutions. Solid Granules fine particles: disintergration deaggregation Solution
 Formulation factors – materials added to the drug during processing can")
Drug Absorption 2) Formulation factors – materials added to the drug during processing can affect the solubilization of the drug. a. Fillers – add bulk to the tablet b. Disintegrators – cause table to break down into granules c. Binders – hold tablet together d. Lubricants – prevent tablet from sticking to machinery Formulation factors - not clinically important if the drug is absorbed effectively and may have important influence on drug absorption for these drugs which are not effectively absorbed in the GI tract - influence drug’s bioavailability.
 Concentration of drug at the absorption site Passive diffusion Driving force")
Drug Absorption 3) Concentration of drug at the absorption site Passive diffusion Driving force – the concentration gradient. The higher the concentration of the drug, the faster the rate of absorption.
 Blood flow at the absorption site - maintain concentration gradient Membrane")
Drug Absorption 4) Blood flow at the absorption site - maintain concentration gradient Membrane Blood
 Surface area of absorption small intestine")
Drug Absorption 5) Surface area of absorption small intestine
 Route of administration GI tract – first pass effect")
Drug Absorption 6) Route of administration GI tract – first pass effect
 Gastric emptying small intestine – primary site of drug absorption Anything")
Drug Absorption 7) Gastric emptying small intestine – primary site of drug absorption Anything that delays/accelerates gastric emptying will decrease/increase drug absorption. For all drugs - acidic, basic or neutral substances.
 Food High fat food – delay gastric emptying – slow absorption")
Drug Absorption 8) Food High fat food – delay gastric emptying – slow absorption
 Intestinal motility – depends on whether the drug is completely absorbed")
Drug Absorption 9) Intestinal motility – depends on whether the drug is completely absorbed under normal condition. a. Completely absorbed early upon entry into the small intestine, increasing intestinal motility will not significantly affect absorption. b. Not completely absorbed before entry into the small intestine, increasing/decreasing intestinal motility will slow down/facilitate drug absorption.
 Metabolism of drug by GI tract a. Drug metabolizing enzymes in")
Drug Absorption 10) Metabolism of drug by GI tract a. Drug metabolizing enzymes in the GI tract b. Proteases in the GI tract c. Microbes in the GI tract - metabolize certain drugs - Drug metabolites are not usually absorbed.

Absorption: Bioavailability – Bioavailability: fraction of oral dose that appears in systemic circulation. • Unless given as liquid, drug must be released by: - Disruption of coating or capsule - Disintegration of tablet - Dispersion throughout stomach or G. I. tract - Dissolution in gastrointestinal fluid
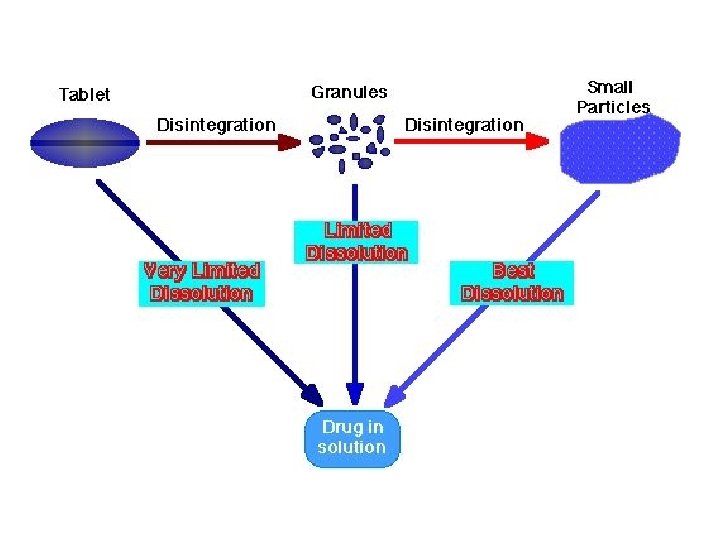

Absorption: Bioavailability

Pharmacokinetics: Drug Distribution • The dispersion or dissemination of substances throughout the fluids e. g. plasma, intracellular fluids and tissues of the body. • 2 drug forms: - free (pharmacologically active), can cross cell membranes - bound to plasma proteins (pharmacologically inactive)

Pharmacokinetics: Drug Distribution Factors affecting distribution: - Plasma proteins - Blood flow to tissues - Specialized barriers - p. H differences between plasma and tissue compartments - Lipid solubility vs. water solubility
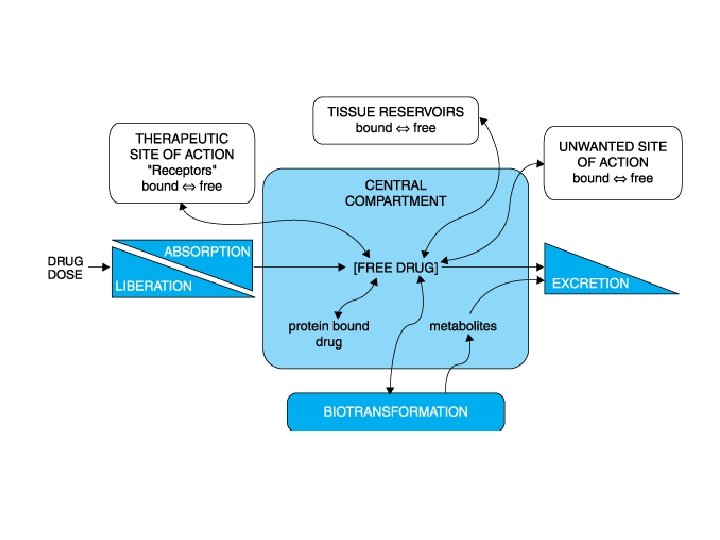
: A hypothetical volume of fluid into which the")
Drug Distribution Volume of distribution (Vd): A hypothetical volume of fluid into which the drug is distributed. - Water compartments in the body - three functionally distinct water compartments – plasma, interstitial fluid and intracellular fluid. Total body water 60% of body weight 60% x 70 kg = 42 liters Intracellular fluid 40% of body weight 28 liters Extracellular fluid 20% of body weight 14 liters Plasma 6% of body weight 4 liters Interstitial volume 14% of body weight 10 liters

Drug Distribution Plasma compartment: Large molecular weight or binds extensively to plasma proteins - trapped in the plasma compartment – Vd = 4 liters. Extracellular fluid: Low molecular weight, Plasma 4 L Intracellular 28 L Interstitial fluid 10 L hydrophilic - able to move through fenestrae into the interstitial fluid, but cannot move across the cell membranes into the intracellular fluid sum of the plasma water and interstitial fluid – Vd = 14 liters. Total body water: Low molecular weight, hydrophobic - move into the interstitial fluid and the intracellular fluid – Vd = 42 liters.

Drug Distribution

Drug Distribution Regional blood flow – unequal distribution of cardiac output Perfusion rate: blood flow to tissue mass ratio Higher: heart, kidney, liver, lung and brain Moderate: muscle and skin Low: adipose tissue The perfusion rate affects the rate at which a drug reaches the equilibrium in the extracellular fluid of a particular tissue. The greater the blood flow, the more rapid the drug distribution from plasma into interstitial fluid. Therefore, a drug will appear in the interstitial fluid of liver, kidney and brain more rapidly than it will in muscle and skin.

Drug Distribution Capillary permeability Drug transfer through capillary – filtration a. Capillary structure: Capillary size and fenestrae size Liver: larger fenestrae - greater filtration potential Brain: smaller fenestrae – lower capillary permeability Liver – slit junction Brain – tight junction -blood-brain barrier
 To penetrate CNS")
Drug Distribution Specialized barriers -Blood-brain barrier (cell layer and basement membrane) To penetrate CNS drug must cross BBB, which consists of epithelial and basement membrane cells Are no pores, gaps etc to allow easy penetration of drugs Drug penetration of BBB relates to lipid solubility and ionization Highly lipid soluble non-ionized drugs easily penetrate BBB to access cerebrospinal fluid e. g. thiopental Highly lipid insoluble and/or ionized drugs in general do not cross BBB and do not affect CNS e. g. hexamethomium -Placenta Drugs pass via simple diffusion governed by lipid solubility ↑ lipid solubility = ↑ drug uptake by fetus Most drugs taken by mother reach the fetus e. g. alcohol

Drug Distribution Blood-brain barrier Liver Brain Large fenestrae Slit junction Drugs Tight junction Small fenestrae Lipid soluble drugs Passive diffusion Carrier-mediated transport Endothelial cells MEMBRANE

Drug Distribution Capillary permeability Drug transfer through capillary – filtration a. Capillary structure: b. Chemical nature of the drug: Sizes of the drug Drug structure: Hydrophobic drugs: passive diffusion – blood flow Hydrophilic drugs – fenestrae - filtration

Drug Distribution Rate of transfer from interstitial fluid into tissues Passive diffusion, active transport and endocytosis. Passive diffusion - the most common and quickest means Blood – plasma Interstitial fluid

Drug Distribution Binding to plasma proteins - reversible Capillary endothelium cells Blood Interstitial fluid Cells and tissues

Drug Distribution Consequence of drug binding to plasma proteins: Cannot go to its receptor at the site of action Cannot be distributed to body tissues Cannot be metabolized by enzymes Cannot be excreted from the body

Drug Distribution - Bound drugs are pharmacologically inactive. - Drug binding to plasma protein will delay the onset of drug action. - Drug binding to plasma proteins will decrease the intensity of drug action.

Drug Distribution Drug binding to plasma proteins may prolong drug action. Reservoir of non-metabolized drug in the body Surmin – trypanosomiasis – A single IV injection may be effective for three months. Warfarin – 97% bound to plasma proteins and 3% free.

Drug Distribution Types of plasma proteins: Albumin: • The primary serum protein responsible for drug binding • 68 k. D • The strongest affinity for weak acid and hydrophobic drugs. • 1 or 2 selective high affinity binding sites for week acidic drugs.

Drug Distribution Types of plasma proteins: Lipoproteins: • Lipid-soluble drugs • The binding capacity is dependent on their lipid content. • Binding ability of lipoproteins is VLDL > HDL. • Patient – more free drug available for absorption in patients with high HDL than patients with high LDL.

Drug Distribution Types of plasma proteins: alpha 1 -acid glycoprotein: • • Alpha 1 - globulin 44 KD One high affinity binding site and binds only basic drugs Plasma concentration - inducible by acute injury, trauma, and stress. • The half time: 5. 5 days. • Patient with trauma taking a basic drug – side effect

More plasma proteins Less free drug available
 Amount of time required for the concentration of")
Drug Distribution Plasma Half-life (t 1/2) Amount of time required for the concentration of a drug to fall to one half of its blood level -When half-life is short drug is quickly removed and duration of action is short e. g. t 1/2 = 1 h drug mostly gone in 4 -5 hours -When drug half-life is long drug slowly removed from body and duration of action is long e. g. t 1/2 = 60 h drug needs 300 h to be mostly gone from body First order kinetics t 1/2 100% t 1/2 50% t 1/2 25% t 1/2 12. 5% t 1/2 6. 25% 3. 13%

Drug Metabolism • Metabolism: Irreversible biochemical transformation of drug into metabolites to increase excretion from the body via the kidney - - Metabolism/Biotransformation occurs mainly in the liver Usually drug is converted to a more water soluble compound Activity of metabolite may be different from parent compound Metabolite usually more polar (ionized) Metabolite usually less lipid soluble Renal tubular absorption of metabolite Metabolites less likely to bind to plasma proteins Metabolites less likely to be stored in fat

Drug Metabolism The liver is the dominant organ in drug metabolism Organ Relative activity (%) Liver Lung Kidney Intestine Placenta Adrenal Akin Leukocytes Spleen Eye brain 100 20 -30 8 6 5 2 1 lower
 Active drug inactive metabolite, most common metabolic")
Drug Metabolism Mechanisms of drug metabolism 1) Active drug inactive metabolite, most common metabolic reaction e. g. doxycyline 2) Inactive drug active drug, prodrug converted to active drug e. g. acyclovir 3) Active drug active metabolite, adds another step before excretion and prolongs drugs actions e. g. diazepam to active metabolite desmethyldiazepam

Drug Metabolism • First pass effect – Drugs taken orally pass through the liver before they get to the systemic circulation. – During first pass through the liver, drug is removed by metabolism or hepatobiliary secretion. – Phase I, oxidation, hydrolysis and reduction (non-synthetic reactions) – Phase II, conjugation (synthetic reactions) – Forms easily excreted polar compound If no functional group Drug Phase I metabolite Phase II If drug has functional group Drug Phase II excretion

Drug Metabolism Phase I metabolism -Metabolizes drugs to creates sites for phase II metabolism 1) Oxidation (adds O) typically via simple addition of O or hydroxylation (adds H and O). Mediated predominantly via microsomal endoplasmic reticulum cytochrome P 450 liver enzymes. Kidney and nervous tissue enzymes can also oxidize compounds 2) Reduction (gain of electron or H). Mediated by P 450 enzymes 3)Hydrolysis (addition of water). Performed by hydrolytic enzymes called plasma esterases e. g. plasma cholinesterase

Drug Metabolism I. Types of non-synthetic reactions 1. Oxidation reactions A. A direct insertion of a hydroxyl functional group into the drug molecule B. Mostly by cytochrome P 450 C. Almost exclusively in the ER D. Broad specificity of cytochrome P 450 – multiple isoforms The hepatic microsomal cytochrome P 450 -dependent electron transfer chain NADPH NADP+ NADPH Cytpchrome P 450 reductase RH – drug substance P 450 RH + O 2 ROH + H 2 O

Drug Metabolism I. Types of non-synthetic reactions 1. Oxidation reactions A. B. C. D. E. F. A direct insertion of a hydroxyl functional group into the drug molecule Mostly by cytochrome P 450 Almost exclusively in the ER Broad specificity of cytochrome P 450 – multiple isoforms Types of microsomal oxidations 1) Aromatic or ring hydroxylation 2) Aliphatic hydroxylation 3) Epoxidation 4) N-, O-, and S-dealkylations 5) N-hydroxylation (not P 450) 6) N-oxidation (not P 450) 7) Oxidative deamination 8) Sulfoxide formation 9) Desulfuration Non-microsomal oxidation 1) Alcohol dehydrogenase 2) Aldehyde dehydrogenase 3) Xanthine oxidase 4) Tyrosine hydroxylase 5) Monoamine oxidase

Drug Metabolism I. Types of non-synthetic reactions 1. Oxidation reactions A. B. C. D. E. F. A direct insertion of a hydroxyl functional group into the drug molecule Mostly by cytochrome P 450 Almost exclusively in the ER Broad specificity of cytochrome P 450 – multiple isoforms Types of microsomal oxidations 1) Aromatic or ring hydroxylation 2) Aliphatic hydroxylation 3) Epoxidation 4) N-, O-, and S-dealkylations 5) N-hydroxylation (not P 450) 6) N-oxidation (not P 450) 7) Oxidative deamination 8) Sulfoxide formation 9) Desulfuration Non-microsomal oxidation 1) Alcohol dehydrogenase 2) Aldehyde dehydrogenase 3) Xanthine oxidase 4) Tyrosine hydroxylase 5) Monoamine oxidase

Drug Metabolism Aromatic hydroxylation

Drug Metabolism Aromatic hydroxylation

Drug Metabolism Aliphatic hydroxylation

Drug Metabolism Epoxidation

Drug Metabolism Oxidative deamination

Drug Metabolism I. Types of non-synthetic reactions 2. Reduction reactions - gain of electron or H A. Catalyzed by reductases B. Reductases - found in microsomes, the cytosol and microorganisms in the gut

Drug Metabolism I. Types of non-synthetic reactions 1. Oxidation reactions – mostly by cytochrome P 450 2. Reduction 3. Hydrolysis A. Breaking compounds with the addition of water B. Primarily occur in the liver, kidney and in the plasma C. Esterase – carry out the major hydrolysis reactions D. Amidase and expoxide hydrolases

Drug Metabolism Hydrolysis catalyzed by esterases Addition of H 2 O

Drug Metabolism Hydrolysis catalyzed by amidases Addition of H 2 O

Drug Metabolism I. Non-synthetic reactions II. Synthetic reactions – conjugation reactions couples agent to existing (or phase I formed) conjugation site on drug/metabolite Involves addition to functional groups including ethers, alcohols, aromatic amines on drug metabolite • Glucuronidation - the most common reaction, occurs in the liver, Glucaroninc acid combines with -OH, -SH, -COOH, -CONH groups to create glucuronide metabolites • Sulfate conjugation - the second important reaction, catalyzed by sulfotransferases in the cytoplasma of the liver and other organs • Acetylation - catalyzed by N-acetyltransferases. Acetic acid combines with NH 2, -CONH 2 groups and aminoacids to give acetylated derivatives • Methylation - catalyzed by methyltransferases in the cytoplasma or ER • Glutathione conjugation - Glutathione combines with -nitrate, epoxide and sulphate groups to create glutathione conjugates • Amino acid conjugation - add naturally occurring amino acids prior to secretion

Drug Metabolism Two pathways of conjugation reactions

Drug Metabolism Factors affecting drug metabolism 1. Age 2. Nutrition and diet 3. Enzyme induction and inhibition by foreign compounds Induction Increase amount/activity of P-450 enzymes Many different compounds are able to alter isoenzyme activity P-450 drug metabolism and drug effect e, g, Phenobaritol stimulates CYP P-450 metabolisim of warfarin and reduces warfarin effect Inhibition Decreases amount/activity of P-450 enzymes P-450 drug metabolism and drug effect

Drug Excretion • Excretion: The elimination of the substances from the body unchanged or as a metabolite. 1. Sites for drug excretion: 1) 2) 3) 4) 5) Kidney - Urine Liver – Bile Skin Lung Milk

Drug Excretion 1. Renal excretion Glomerular filtration Drug/metabolite filtered via glomeruli and concentrated in renal tubular fluid and excreted in urine. Protein bound drug remains in systemic circulation. • • • Drugs from glomerulers into the renal tubules Pressure – blood flow MW cut off = 5000 7500 – restricted Lipid soluble drugs – also by passive diffusion
 Glomerular filtration 2) Active secretion • Two active")
Drug Excretion 2. Renal excretion 1) Glomerular filtration 2) Active secretion • Two active transport systems: Organic acids Organic bases • Relatively non-specific • Unidirection – accumulation and excretion
 Glomerular filtration 2) Active secretion 3) Passive reabsorption")
Drug Excretion 2. Renal excretion 1) Glomerular filtration 2) Active secretion 3) Passive reabsorption - Affected by urinary p. H • Unionized, lipid soluble metabolites are reabsorbed • Ionized, lipid-insoluble metabolites excreted in urine More ionization – more secretion

Forced alkaline diuresis Phenobarbital – weak acid Renal tubule p. H = 5 – 8 -Alkaline urine = increased weak acid excretion, reduced weak base excretion -Acidic urine = decreased weak acid excretion, increased weak base excretion Bicarbonate–increase p. H–ionized–faster excretion Ammonium chloride – decrease p. H

Drug Excretion Kidneys 3. Secretion from the liver: Liver - Bile – intestine • Liver: Metabolizing enzymes Active transport systems – bile capillaries • Lipid insoluble or ionized drugs – excretion • Lipid soluble – reabsorption from intestine to bile – transport back to the liver Enterohepatic cycling: Prolong drug action Conserve endogenous substances – VD 3, B 12, folic acid, estrogens.

Drug Excretion 4. Pulmonary excretion Gasses and volatile liquids Simple diffusion from the blood into the airway

Drug Excretion 5. Sweat and saliva Drugs or drug metabolites Passive diffusion Side reaction of the skin

Drug Excretion 6. Milk Passive diffusion Not very important for mother May be important for infant
- Slides: 101