Biological Sequence Determination DNA Robert M Horton Ph

Biological Sequence Determination DNA Robert M. Horton, Ph. D, MS rmhorton@cybertory. org artwork: commons. wikimedia. org protein

 automation, base")
Sequencing context protein RNA DNA technological biological old methods classical sequencing (Sanger) automation, base calling, quality scoring shotgun sequencing, assembly, finishing concepts chemistry, enzymes physics, computers contemporary methods: pyrosequencing, CRT, SOLi. D "next generation" applications: resequencing, epigenetics, RNA-Seq “third generation” SMRT, nanopores, etc. microfluidics microfabrication contemplation of the future
 (purifyable) Important Insulin")
Protein Sequencing Why Proteins? Small Digestible (pepsin, trypsin, Chemically distinguishable chymotrypsin) (purifyable) Important Insulin Fred Sanger Nobel prize, 1958

Classes of RNA modified bases (GMe, GMe 2, CMe, T, ψ, UH 2, I, IMe) r. RNA modified bases ( cap with m 7 G , 2'O-methylation ) splicing polyadenylation sn. RNA prokaryotic: 70 S = 50 S (5 S, 23 S) + 30 S (16 S) eukaryotic: 80 S = 60 S (5 S, 5. 8 S, 28 S) + 40 S (18 S) 7 SL RNA of Signal Recognition Particle (SRP) homologous to Alu SINE (11% of human genome) splicosomes (U 1, U 2, U 4, U 5, U 6) sno. RNA t. RNA pre-r. RNA processing (U 3) guide 2'-O-methylation guide pseudouridylation RNAi si. RNA (short interfering RNA) mi. RNA (micro. RNA) post-transcriptional gene silencing 3' UTR, conserved pi. RNA transcriptional silencing of retrotransposons . . . et cetera. . .

DNA Sequencing 1977 The “modern era” of DNA sequencing begins
 February 1977 Two steps: Damage bases specific, partial Cleave")
Chemical Sequencing of DNA (Maxam-Gilbert) February 1977 Two steps: Damage bases specific, partial Cleave backbone Four reactions: A A+G C C+T http: //nobelprize. org/nobel_prizes/chemistry/laureates/1980/gilbert-lecture. pdf
 2', 3'-dideoxy TTP Sanger F, Nicklen S & Coulson")
Chain Termination Sequencing (Sanger Sequencing) 2', 3'-dideoxy TTP Sanger F, Nicklen S & Coulson AR DNA sequencing with chain-terminating inhibitors PNAS 74: 5463 -7, December 1977

Primer Extension Bacterial DNA polymerase I adds nucleotides to the 3' end of primer to complement 5' -overhanging template. Each strand is an ordered sequence with a direction. Arrows indicate 5' to 3' direction (DNA grows biochemically in this direction). (pyrophosphate released)

Sanger sequencing Individual reactions with one d. NTP partially “poisoned” with dideoxynucleotides (dd. ATP, dd. CTP, dd. GTP, dd. TTP) Decades of improvements automated fluorescence four colors one lane dye terminators one reaction capillaries

Automated Sanger sequencing trace base calls quality scores
 p = predicted error probability")
Quality Score q = -10 * log 1 0(p) p = predicted error probability 1/1000 probability of error = q score of 30 uses data quality monitoring assembly consensus finishing criteria
 Shotgun Sequencing (parallel)")
Sequencing Strategy Primer walking (serial) Shotgun Sequencing (parallel)

Universal Primers

Assembly

read length affects assembly
 Cycles of Reversible Termination (Solexa/Illumina) Ligation (ABI SOLi. D) \"Third-Generation\"")
Next-Generation Sequencing Pyrosequencing (454/Roche) Cycles of Reversible Termination (Solexa/Illumina) Ligation (ABI SOLi. D) "Third-Generation" Sequencing SMRT (Pacific Biosciences)
 adenosine 5`-phosulfate ATP sulfurylase +")
pyrosequencing + pyrophosphate APS (released by d. NTP incorporation) adenosine 5`-phosulfate ATP sulfurylase + sulfate ATP

pyrosequencing + ATP O 2 oxygen + luciferin firefly luciferase AMP + pyrophosphate + light + oxyluciferin

pyrosequencing more biochemistry problem solution pyrophosphate recycling apyrase breaks down ATP to AMP + 2 Pi (or wash out solution) luciferase can use d. ATP use an analog suitable for polymerase but not luciferase

pyrosequencing flowgram Ronaghi M. Genome Res 11: 3 -11, 2001

Emulsion PCR water droplet in oil one primer bound to solid bead individual template molecule

Emulsion PCR DNA anchored to bead all comes from the same template molecule "polony" = "PCR colony"
 p. H change (\"Ion Torrent\")")
pyrosequencing Alternatives to chemiluminescence heat (“thermosequencing”) p. H change ("Ion Torrent")

Cycles of Reversible Termination Illumina/Solexa Helicos Illumina Metzker M. Sequencing Technologies - The Next Generation. Nature Reviews Genetics 11: 31 -46, 2010.

Short Read Alignment
 + 33);")
FASTQ Format maq. sourceforge. net/fastq. shtml $q = chr(($Q<=93? $Q : 93) + 33); 0 60 $Q = ord($q) - 33; !"#$%&'()*+, -. /0123456789: ; <=>? @ABCDEFGHIJKLMNOPQRSTUVWXYZ[]
")
Paired End Tags Mme I TCCRAC (20/18)

Illumina Genome Analyzer Library Preparation

Illumina Genome Analyzer Bridge Amplification forms "Polonies"

Illumina Genome Analyzer Cycles of Reversible Termination
 Complete Genomics Polonator (Church Lab)")
Ligation-based Sequencing SOLi. D (ABI) Complete Genomics Polonator (Church Lab)

SOLi. D Sequencing by Oligonucleotide Ligation and Detection 3'- ATNNN~ZZZ*-5' artwork is from the pamphlet Dibase Sequencing and Color Space Analysis
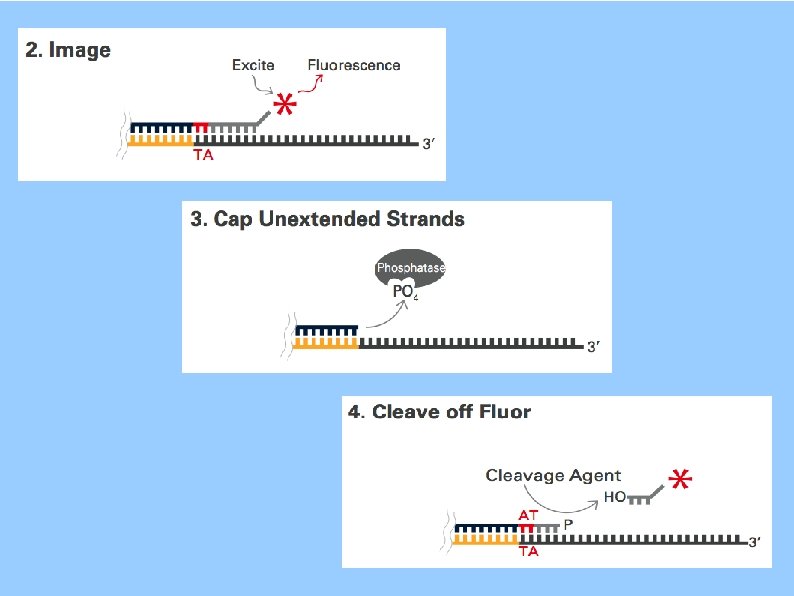
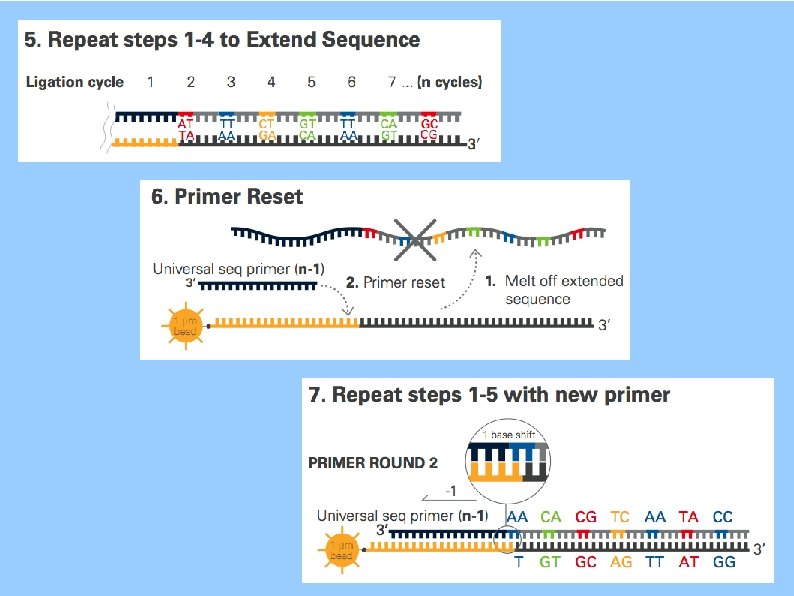

SOLi. D

SOLi. D: Dibase Encoding AT CG GC TA AC CA GT TG AA CC GG TT AG CT GA TC

SOLi. D: Dibase Encoding base space color space Each color sequence can represent four different base sequences. The base sequence is one unit longer than the color sequence. You need to know one base to tell which sequence is represented.

SOLi. D: Dibase Encoding SNP causes two color changes single color change is probably an error
 Sequencing High throughput Parallelism (small reactions) Speed (immediate results) Long reads")
Single-Molecule, Real-Time (SMRT) Sequencing High throughput Parallelism (small reactions) Speed (immediate results) Long reads Read individual templates from mixtures Haplotyping

SMRT Sequencing 41

Simulated SMRT Sequencing Data

Platform Comparisons Xu M, Fujita D, and Hanagata N. Perspectives and Challenges of Emerging Single-Molecule DNA Sequencing Technologies. Small 5(23): 2638– 2649, 2009
 ionic current blockage")
Other Technologies Mass spectrometry TEM STM nanonozzle probes nanopores (protein, graphene) ionic current blockage transverse tunneling currents exonuclease

Targeted Exome Capture nimblegen. com

Bonus Slides

Selenocysteine t. RNA

Omics transcriptome exome kinome
 \"minus\": polymerase stops at missing base \"plus\": T")
“Plus and Minus” Method (circa 1975) "minus": polymerase stops at missing base "plus": T 4 DNA polymerase 3' exonuclease stalled by d. NTP Sanger F, Coulson AR. J Mol Biol. 94(3): 441 -8, 1975

pyrosequencing Animation: http: //www. pyrosequencing. com/Dyn. Page. aspx? id=7454

Bioinformatics Classics Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443 -453, 1970. Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol 147: 195 -197, 1981.

Automated Base Calling 1. identify idealized peak locations assume locally even spacing 2. find observed peaks 3. match observed to expected omit and split as necessary 4. add "good" unmatched peaks

Error Probabilities predictive does not require knowing actual sequence valid the set of bases assigned to probability p should have an actual error rate of p discriminating helps to distinguish correct vs. incorrect base calls 1, 000 base calls with 1, 0000 errors (p = 0. 01) better if we can break it into two 500, 000 sets: p=0. 018 in one set (9000 errors) p=0. 002 in second set (1000 errors)

Error Probability Calibration 'Given a set of parameters and a training set of reads for which it is known which base-calls are correct and which are errors, find a way of associating parameter values to error probabilities that has (near) maximum discrimination power for small r. '

Phred Quality Score Parameters Empirical. Small values tend to correspond to more accurate base-calls. Window-based parameters smooth out error probabilities. 1. Peak spacing (7 peak window) ● largest / smallest peak-to-peak spacing 2. Uncalled/called ratio (7 peak window) ● amplitude of largest uncalled / smallest called peak 3. Uncalled/called ratio (3 peak window) 4. Peak resolution ● -1 * # bases to the next unresolved base

Lookup Table Production Select a range of 50 threshold values for each of the 4 parameters. For each 4 -tuple of parameter thresholds (504=6, 250, 000): find the set of bases defined by these thresholds compute empirical error rates The parameter set with the lowest error rate goes into the table. These 50 values are chosen so that each increment contains approximately the same number of bases in the training set. if multiple 4 -tuples give the same rate, choose the largest set These bases are removed, and the process is repeated until all bases are represented in the table.
 glycosylation (glycoproteins) mucin, cellular interaction, structural N-linked hydroxylysine in collagen")
Post-translational Modification (or co-translational) glycosylation (glycoproteins) mucin, cellular interaction, structural N-linked hydroxylysine in collagen covalently bound enzyme cofactors thyroid hormone hydroxylation FAD, biotin, etc ubiquitination methylation isoprenylation phosphorylation acetylation (acetate, CH 3 CO 2− ) myristoylation (myristate, a C 14 fatty acid) palmitoylation (palmitate, a C 16 fatty acid) alkylation iodination asparagine serine, threonine, hydroxylysine, hydroxyproline acylation (at O, N, or S) O-linked signal transduction ADP-ribosylation signal transduction cholera toxin . . . and many more

“Wandering Spot” Method ca. 1970 s RNA or DNA partial digestion 2 D separation Horizontal = base composition Vertical = size This is an RNAse T 1 fragment, so it ends in G Fuke, M. , and Busch, H. Nucleic Acids Res. 4: 339 -352, 1977.

Enzymatic vs Chemical Partial Cleavage of RNA Sequence-specific RNases Phy M: A+U A: pyrimidine-specific (C+U) U 2: A or A+G T 1: degrades after G residues V 1: degrades paired bases Peattie DA. PNAS 76: 1760 -1764, 1979. enzymatic chemical
 methyl guanine (GMe) pseudouridine (ψ) dimethylguanine(GMe 2) dihydrouridine")
Modified Nucleotides in t. RNA (post-transcriptional) methyl guanine (GMe) pseudouridine (ψ) dimethylguanine(GMe 2) dihydrouridine (UH 2) methylcytosine (Me) inosine (I) ribothymine (T) methylinosine (IMe)
 Unambiguous A, C, G, T, U 2 -fold degenerate M")
Nucleotide Ambiguity Codes (IUPAC) Unambiguous A, C, G, T, U 2 -fold degenerate M = A or C R = A or G (pu. Rine) W = A or T (Weak) S = C or G (Strong) Y = C or T (p. Yrimidine) K = G or T 3 -fold degenerate V = A, C or G (not T) H = A, C or T (not G) D = A, G or T (not C) B = C, G or T (not A) 4 -fold degenerate X = A, C, G or T N = A, C, G or T
")
Automated Base Calling Phred third-party base caller with better accuracy than ABI's open source(ish) Ewing B, Hillier L, Wendl MC, Green P. Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Res. 8: 175 -185, 1998 Ewing B and Green P. Base-Calling of Automated Sequencer Traces Using Phred. II. Error Probabilities. Genome Res. 8: 186 -194, 1998

Shotgun Sequencing Staden R. A strategy of DNA sequencing employing computer programs, Nucleic Acids Research 7: 2601 -2610, 1979 “With modern fast sequencing techniques and suitable computer programs it is now possible to sequence whole genomes without the need of restriction maps. This paper describes computer programs that can be used to order both sequence gel readings and clones. A method of coding for uncertainties in gel readings is described. These programs are available on request. ” “The whole of the DNA to be sequenced is shotgunned into a suitable vector and cloned. Ideally the cloned fragments would be of at least 200 bases in length. The clones are then sequenced and the computer used to collate the data. Collation involves searching for overlaps in the data. ”

2 D gel electrophoresis

cybertory. org/exercises/primer. Design
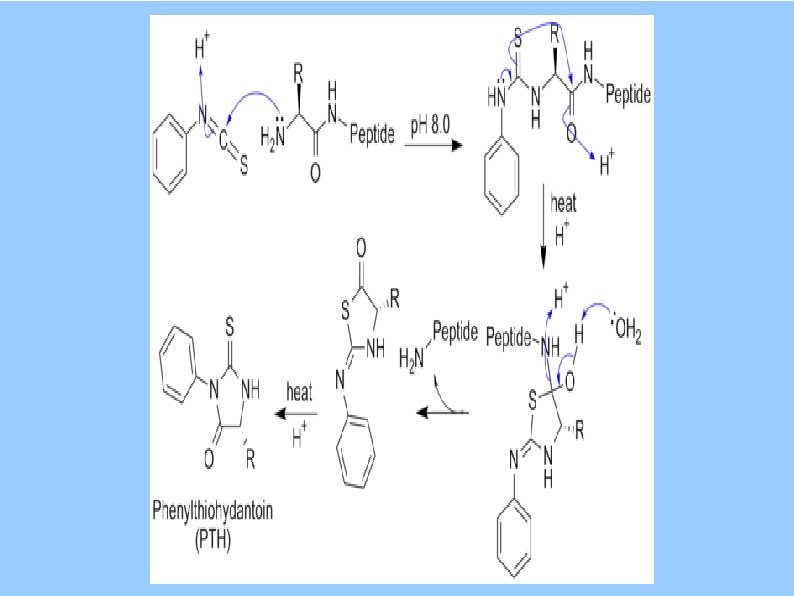

Protein Sequencing Edman Degradation phenylisothiocyanate invented ca. 1950 s automated ca. 1973 proceeds from N-terminus read 50 -70 aa http: //en. wikipedia. org/wiki/Edman_degradation A few amino acids can ID a spot on 2 D gel Mass Spectrometry Precise determination of molecular weights of peptides
 modified from Wikimedia commons")
(Sec) modified from Wikimedia commons
- Slides: 68