Biological Databases Part II Secondary Databases It is

Biological Databases Part II

Secondary Databases • It is also known as curated database. • Databases consist of data derived from the analysis of primary data such as sequences. • Contains results of analysis of primary databases and significant data in form of conserved sequences, signature sequences, active site residues of proteins.

Secondary Databases • Secondary Databases: PROSITE, BLOCKS, PRINTS, Pfam • Secondary Composite or Structural : CATH, SCOP • Metabolic Pathways: KEGG

Multiple Sequence Alignment • A linear comparison of nucleic acids or amino acid sequences in which insertions are made in order to bring equivalent positions in adjacent sequences into correct register. • Alignments are the basis of sequence analysis methods and are used to pinpoint the occurrence of conserved motifs.

Multiple Sequence Alignment

Motif • A consecutive string of amino acids in a protein sequence whose general character is repeated or conserved in all sequences in a multiple alignment at a particular position. • Motifs are of interest because they may correspond to structural or functional elements within the sequences they characterise.

Motif

Regular Expression • A single consensus expression derived from a conserved region of a sequence alignment and used as a characteristic signature of family membership.

Rule • A short regular expression, typically 4 -6 residues long, used to identify generic – non family specific pattern in protein sequences. • Rules tend to be used to encode particular functional sites like phosphorylation, hydroxylation.

Fingerprint • A group of ungapped motifs excised from a sequence alignment and used to build a characteristic signature of family membership by means of iterative searching of a primary database.

BLOCKS • An ungapped, aligned motif consisting of sequence segments that are clustered to reduce multiple contributions from groups of highly similar or identical sequences.

Hidden Markov Model • A probabilistic model consisting of a number of interconnecting states. • Encodes full domain alignments. • They are essentially linear chains of match, delete or insert state: a match denotes a conserved column in an alignment; an insert state allows insertions relative to match states; and delete states allow match position to be skipped.

Hidden Markov Model

PROSITE • First database to be developed. • Maintained by Swiss Institute of Bioinformatics. • Motifs are encoded as Regular Expression referred to as patterns. • Sequence information within motifs is reduced as single consensus expressions. • Resulting seed pattern are used to search SWISS-PROT.
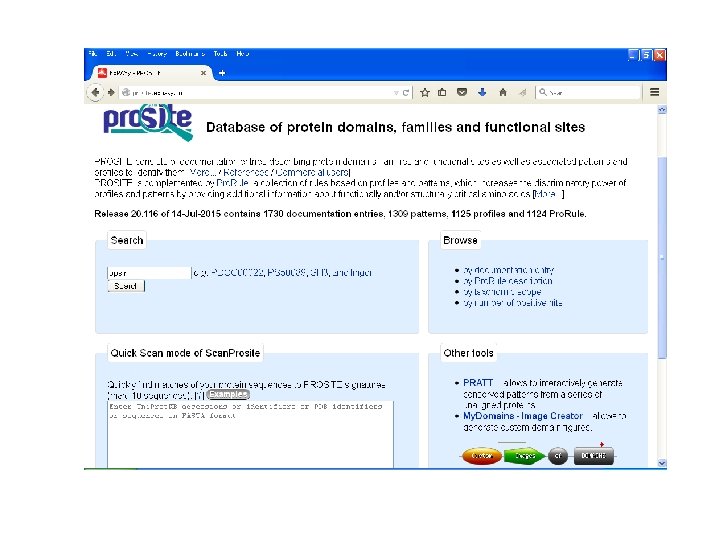

PROSITE

PROSITE

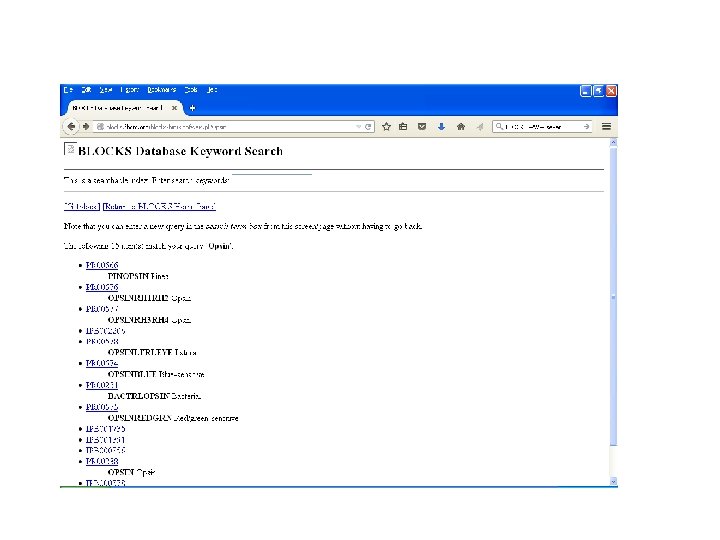
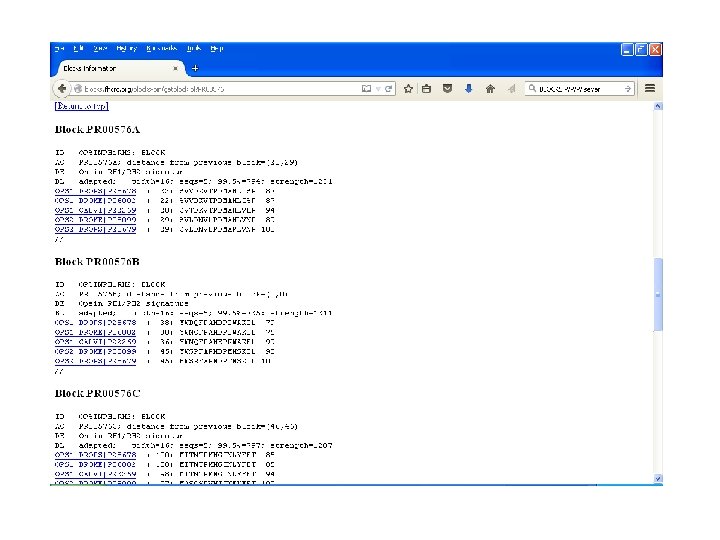
BLOCKS • Created a multiple- motif databased on protein families in PROSITE. • Developed at Fred Hutchinson Cancer Research Center (FHCRC)- Seattle. • BLOCKs are created by automatically detecting the most highly conserved regions of each protein family. • Resulting blocks calibrate against SWISS-PROT for likelihood match.

BLOCKS

PRINTS • Uses Fingerprints as diagnostic signature of family membership. • Developed by Dept. of Biochemistry and Molecular Biology, UCL. • Uses OWL, SWISS-PROT, SP+Tr. EMBL • Uses iterative method to match motifs in the fingerprint.

PRINTS
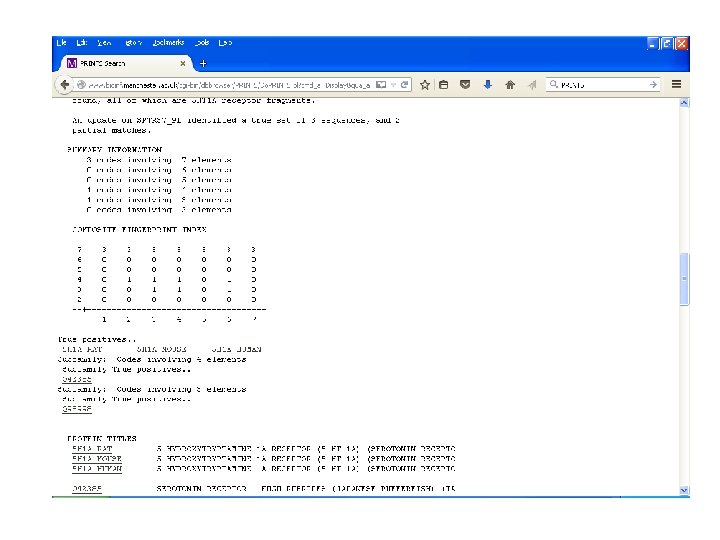

PRINTS
. • Maintained by Sanger Centre.")
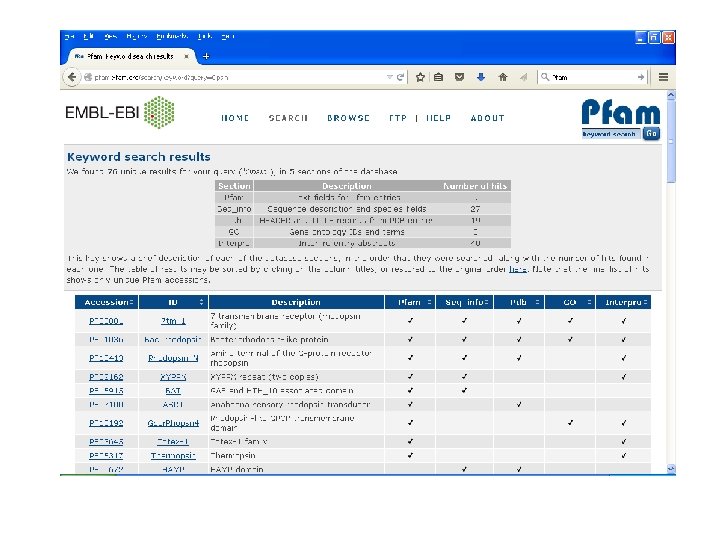
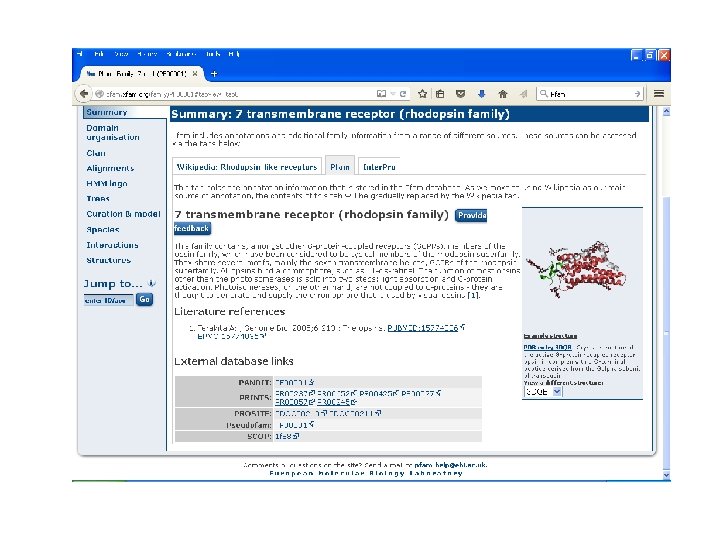
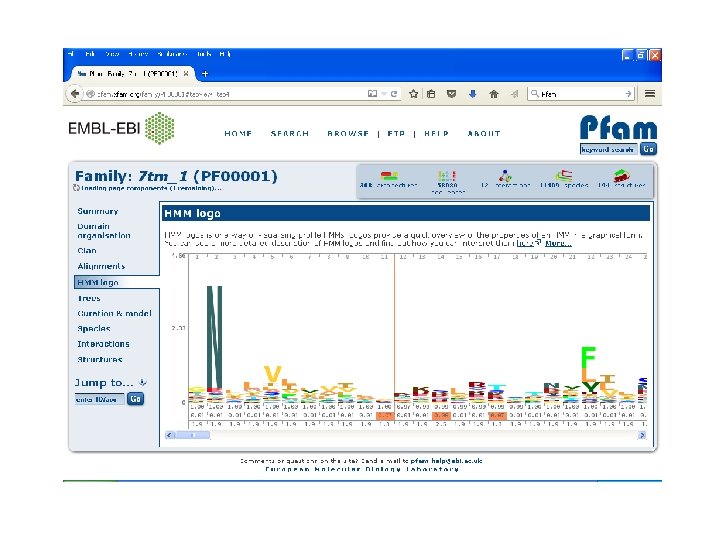
Pfam • Encodes alignments in Hidden Markov Models (HMM). • Maintained by Sanger Centre. • Uses automatic clustering of SWISS-PROT. • Uses full alignments to generate high quality seed alignments to build HMM. • Its iterative method. • Gives one of the best results.

Pfam

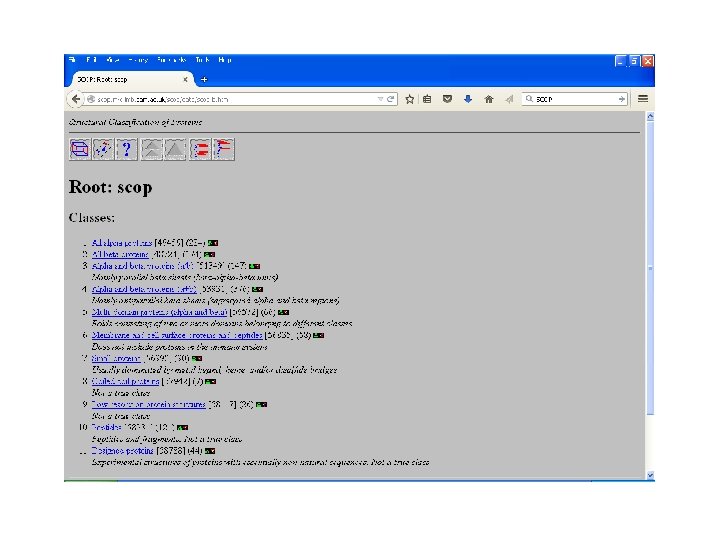
SCOP • Structural Classification Of Proteinsmaintained by MRC Laboratory. • Classifies proteins as: – Family: with evolutionary relationship, more 30% homology. – Superfamily: based on structural and functional characteristics- common evolutionary ancestor. – Fold: having common fold , same topology

SCOP
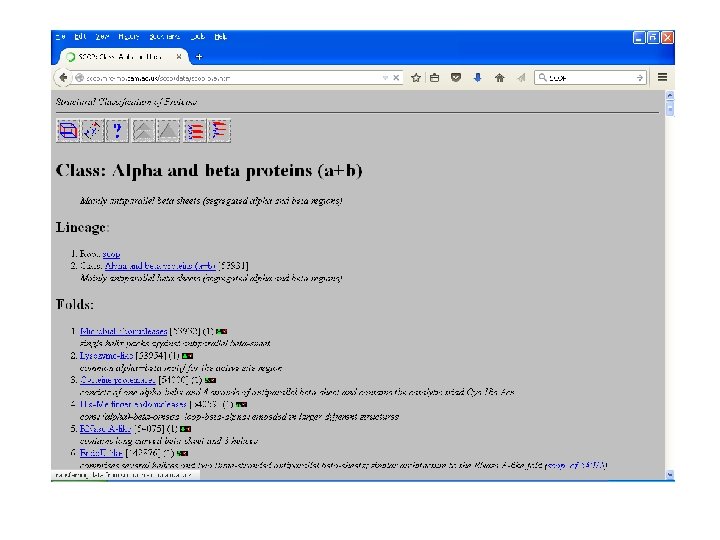
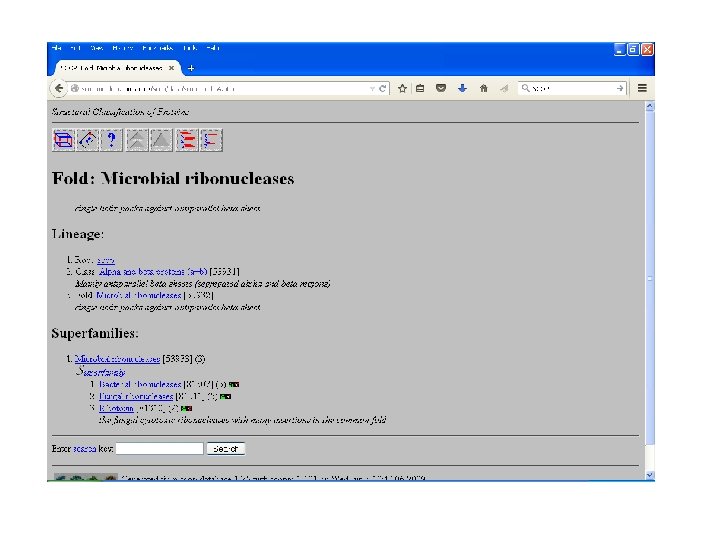
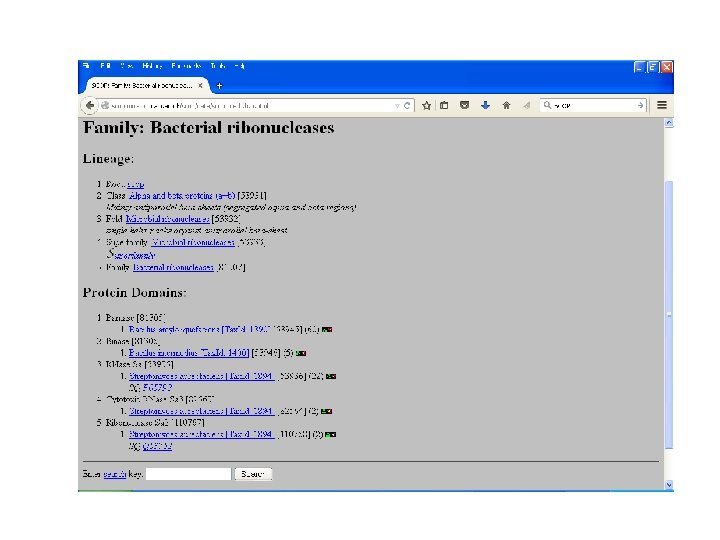
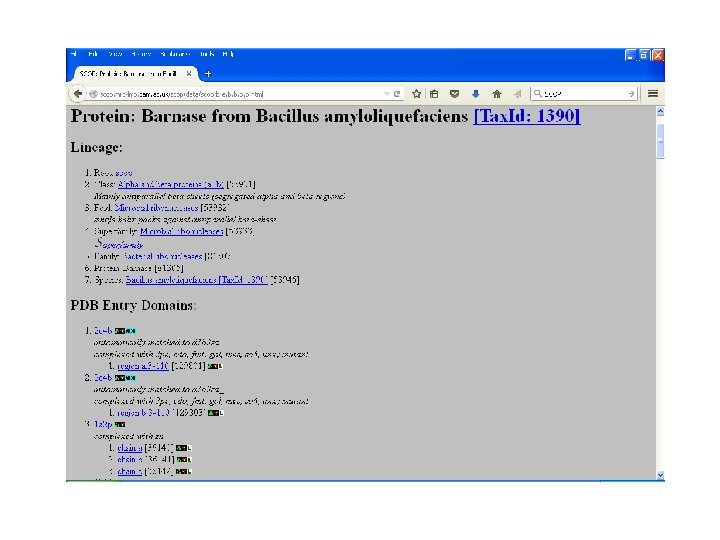

CATH • Class: gross secondary structure like α- helix, βsheets etc. • Architecture: gross arrangement of secondary structures like barrel, sandwich, roll. • Topology: overall shape and connectivity of secondary structure. • Homology: sequence identity => 35%, common ancestor. • Sequence: structure within homology group further clustered on the basis of sequence identity- highly similar structures and function. • Maintained by UCL

CATH

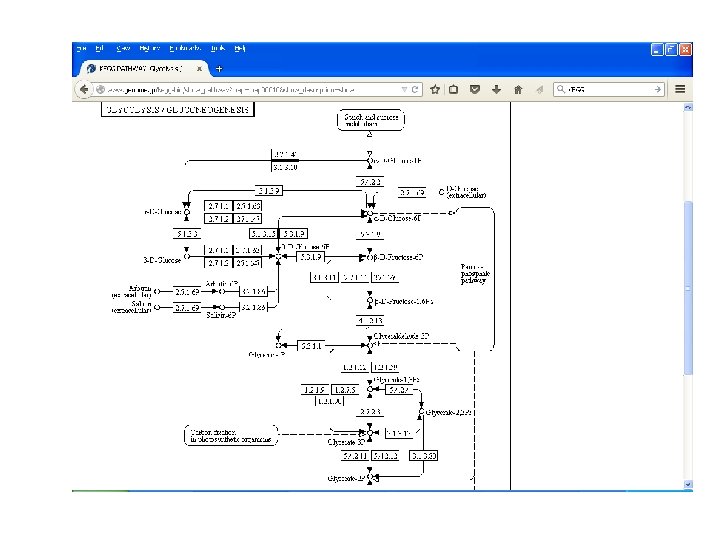
KEGG • Kyoto Encyclopedia of Genes and Genomes • It is a collection of manually drawn KEGG pathway maps representing experimental knowledge on metabolism and various other functions of the cell and the organism. • This has enabled the analysis called KEGG pathway mapping, whereby the gene content in the genome is compared with the KEGG PATHWAY database to examine which pathways and associated functions are likely to be encoded in the genome.

KEGG • • • PATHWAY — pathway maps for cellular and organism functions MODULE — modules or functional units of genes BRITE — hierarchical classifications of biological entities Genomic information GENOME — complete genomes GENES — genes and proteins in the complete genomes ORTHOLOGY — ortholog groups of genes in the complete genomes Chemical information COMPOUND, GLYCAN — chemical compounds and glycans REACTION, RPAIR, RCLASS — chemical reactions ENZYME — enzyme nomenclature Health information DISEASE — human diseases DRUG — approved drugs ENVIRON — crude drugs and health-related substances

KEGG
 is a crystallographic database for the threedimensional")
PDB • The Protein Data Bank (PDB) is a crystallographic database for the threedimensional structural data of large biological molecules, such as proteins and nucleic acids. The data, typically obtained by X-ray crystallography or NMR spectroscopy submitted by biologists and biochemists from around the world, are freely accessible on the Internet via the websites of its member organizations.

PDB

Thank You
- Slides: 52