Bioinformatics Methods Course Multiple Sequence Alignment Burkhard Morgenstern

Bioinformatics Methods Course Multiple Sequence Alignment Burkhard Morgenstern University of Göttingen Institute of Microbiology and Genetics Department of Bioinformatics Göttingen, October/November 2006

Tools for multiple sequence alignment T Y I M R E A Q Y E T C I V M R E A Y E

Tools for multiple sequence alignment T Y I - M R E A Q Y E T C I V M R E A - Y E

Tools for multiple sequence alignment T T Y Y Y C I I M A M V Q M R M E R V E A E Q Q Q A Q Y Y E E E

Tools for multiple sequence alignment T T Y Y Y C – I I V A M M R R Q R E E A A V - Q Q Q Y Y Q Y E E

Tools for multiple sequence alignment T T Y Y C Y – I I V A M M R R Q R E E A A V - Q Q Q Y Y Q Y E E Astronomical Number of possible alignments!

Tools for multiple sequence alignment T T Y Y C Y – I I V A M M M R M Q R E E A E V - Q A Q Q Y Y Q Y E E Astronomical Number of possible alignments!

Tools for multiple sequence alignment T T Y Y C Y – I I V A M M R R Q R E E A A V - Q Q Q Y Y Q Y E E Which one is the best ? ? ?
 What is")
Tools for multiple sequence alignment Questions in development of alignment programs: (1) What is a good alignment? → objective function (`score’) (2) How to find a good alignment? → optimization algorithm First question far more important !
 n What")
Tools for multiple sequence alignment Before defining an objective function (scoring scheme) n What is a biologically good alignment ? ?

Tools for multiple sequence alignment Criteria for alignment quality: 1. 3 D-Structure: align residues at corresponding positions in 3 D structure of protein! 2. Evolution: align residues with common ancestors!

Tools for multiple sequence alignment T T - Y C Y Y I I V A M M M R M Q R E E A E V - Q A Q Q Y Y Q Y E E Alignment hypothesis about sequence evolution Search for most plausible hypothesis!

Tools for multiple sequence alignment Compute for amino acids a and b n Probability pa, b of substitution a → b (or b → a), n Frequency qa of a Define s(a, b) = log (pa, b / qa qb)

Tools for multiple sequence alignment

Tools for multiple sequence alignment Traditional objective functions: Define Score of alignments as n Sum of individual similarity scores s(a, b) n Gap penalty g for each gap in alignment Needleman-Wunsch scoring system (1970) for pairwise alignment (= alignment of two sequences)

T Y W I V T - - L V Example: Score = s(T, T) + s(I, L) + s (V, V) – 2 g

T Y W I V T - - L V Idea: alignment with optimal (maximal) score probably biologically meaningful. Dynamic programming algorithm finds optimal alignment for two sequences efficiently (Needleman and Wunsch, 1970).

Tools for multiple sequence alignment Traditional Objective functions can be generalized to multiple alignment (e. g. sum-of-pair score, tree alignment) Needleman-Wunsch algorithm can also be generalized to find optimal multiple alignment, but: Very time and memory consuming! -> Heuristic algorithm needed, i. e. fast but sub-optimal solution

Tools for multiple sequence alignment Most commonly used heuristic for multiple alignment: Progressive alignment (mid 1980 s)

`Progressive´ Alignment WCEAQTKNGQGWVPSNYITPVN WWRLNDKEGYVPRNLLGLYP AVVIQDNSDIKVVPKAKIIRD YAVESEAHPGSFQPVAALERIN WLNYNETTGERGDFPGTYVEYIGRKKISP

`Progressive´ Alignment WCEAQTKNGQGWVPSNYITPVN WWRLNDKEGYVPRNLLGLYP AVVIQDNSDIKVVPKAKIIRD YAVESEAHPGSFQPVAALERIN WLNYNETTGERGDFPGTYVEYIGRKKISP Guide tree

`Progressive´ Alignment WCEAQTKNGQGWVPSNYITPVN WW--RLNDKEGYVPRNLLGLYPAVVIQDNSDIKVVP--KAKIIRD YAVESEASFQPVAALERIN WLNYNEERGDFPGTYVEYIGRKKISP Profile alignment, “once a gap - always a gap”

`Progressive´ Alignment WCEAQTKNGQGWVPSNYITPVN WW--RLNDKEGYVPRNLLGLYPAVVIQDNSDIKVVP--KAKIIRD YAVESEASVQ--PVAALERIN-----WLN-YNEERGDFPGTYVEYIGRKKISP Profile alignment, “once a gap - always a gap”

`Progressive´ Alignment WCEAQTKNGQGWVPSNYITPVNWW--RLNDKEGYVPRNLLGLYPAVVIQDNSDIKVVP--KAKIIRD YAVESEASVQ--PVAALERIN-----WLN-YNEERGDFPGTYVEYIGRKKISP Profile alignment, “once a gap - always a gap”

`Progressive´ Alignment WCEAQTKNGQGWVPSNYITPVN-------WW--RLNDKEGYVPRNLLGLYP-------AVVIQDNSDIKVVP--KAKIIRD------YAVESEA---SVQ--PVAALERIN-----WLN-YNE---ERGDFPGTYVEYIGRKKISP Profile alignment, “once a gap - always a gap”

CLUSTAL W Most important software program: CLUSTAL W: J. Thompson, T. Gibson, D. Higgins (1994), CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment … Nuc. Acids. Res. 22, 4673 4680 (~ 20. 000 citations in the literature)

Tools for multiple sequence alignment Problems with traditional approach: n Results depend on gap penalty n Heuristic guide tree determines alignment; alignment used for phylogeny reconstruction n Algorithm produces global alignments.

Tools for multiple sequence alignment Problems with traditional approach: But: Many sequence families share only local similarity E. g. sequences share one conserved motif

Local sequence alignment EYENS ERYAS Find common motif in sequences; ignore the rest

Local sequence alignment E-YENS ERYA-S Find common motif in sequences; ignore the rest

Local sequence alignment E-YENS ERYA-S Find common motif in sequences; ignore the rest – Local alignment
")
Gibbs Motive Sampler Local multiple alignment without gaps: C. E. Lawrence et al. (1993) Detecting subtle sequence signals: a Gibbs Sampling Strategy for Multiple Alignment Science, 262, 208 - 214

Traditional alignment approaches: Either global or local methods!

New question: sequence families with multiple local similarities Neither local nor global methods appliccable

New question: sequence families with multiple local similarities Alignment possible if order conserved
, PNAS 93, 12098 -12103 n Combination of")
The DIALIGN approach Morgenstern, Dress, Werner (1996), PNAS 93, 12098 -12103 n Combination of global and local methods n Assemble multiple alignment from gap-free local pair-wise alignments (, , fragments“)

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atc------taatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atc------taatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaa--gagtatcacccctgaataa

The DIALIGN approach atc------taatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaa--gagtatcacc-----cctgaataa

The DIALIGN approach atc------taatagttaaactcccccgtgc-ttag cagtgcgtgtattactaac-----gg-ttcaatcgcg caaa--gagtatcacc-----cctgaataa

The DIALIGN approach Consistency! atc------taatagttaaactcccccgtgc-ttag cagtgcgtgtattactaac-----gg-ttcaatcgcg caaa--gagtatcacc-----cctgaataa

The DIALIGN approach atc------TAATAGTTAaactcccc. CGTGC-TTag cagtgc. GTGTATTACTAAc-----GG-TTCAATcgcg caaa--GAGTATCAcc-----CCTGaa. TTGAATaa

The DIALIGN approach Multiple alignment: atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa
 Calculate all optimal pair-wise alignments")
The DIALIGN approach Multiple alignment: atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaccctgaattgaagagtatcacataa (1) Calculate all optimal pair-wise alignments
 Calculate all optimal pair-wise alignments")
The DIALIGN approach Multiple alignment: atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa (1) Calculate all optimal pair-wise alignments
 Calculate all optimal pair-wise alignments")
The DIALIGN approach Multiple alignment: atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa (1) Calculate all optimal pair-wise alignments

The DIALIGN approach Fragments from optimal pair-wise alignments might be inconsistent

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaa--gagtatcacccctgaataa

The DIALIGN approach atc------taatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaa--gagtatcacccctgaataa

The DIALIGN approach atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaataa

The DIALIGN approach Score of alignment: n Define weight score for fragments based on probability of random occurrence n Score of alignment = sum of weight scores of fragments Goal: find consistent set of fragments with maximum total weight

The DIALIGN approach Advantages of segment-based approach: n Program can produce global and local alignments! n Sequence families alignable that cannot be aligned with standard methods
, T-Coffee: A novel algorithm for multiple")
T-COFFEE C. Notredame, D. Higgins, J. Heringa (2000), T-Coffee: A novel algorithm for multiple sequence alignment, J. Mol. Biol.
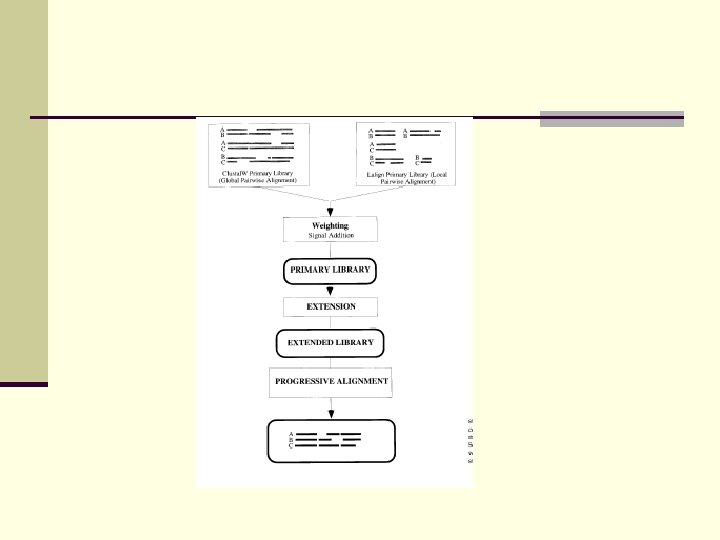

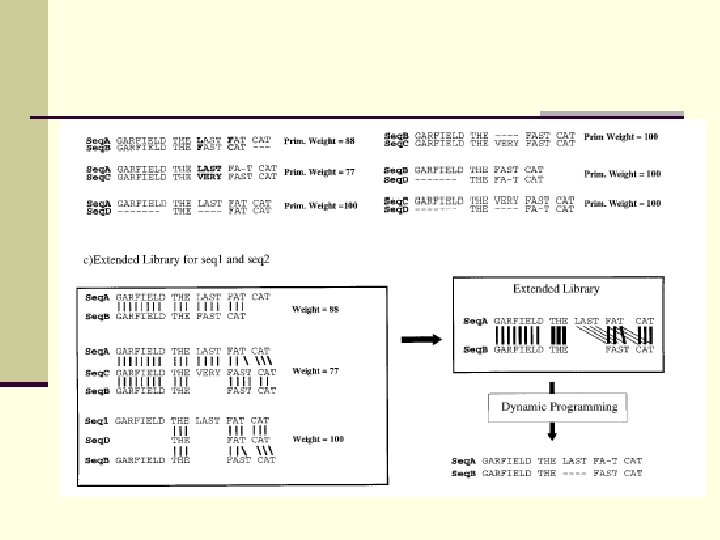

T-COFFEE n Less sensitive to spurious pairwise similarities n Can handle local homologies better than CLUSTAL

T-COFFEE Idea: 1. Build library of pairwise alignments 2. Alignment from seq i, j and seq j, k supports alignmetn from seq i, k.

Evaluation of multi-alignment methods Alignment evaluation by comparison to trusted benchmark alignments. `True’ alignment known by information about structure or evolution.
:")
Evaluation of multi-alignment methods For protein alignment: n M. Mc. Clure et al. (1994): 4 protein families, known functional sites n J. Thompson et al. (1999): Benchmark data base, 130 known 3 D structures (BAli. BASE) n T. Lassmann & E. Sonnhammer (2002): BAli. BASE + simulated evolution (ROSE)

Evaluation of multi-alignment methods

Evaluation of multi-alignment methods 1 abo. A 1 ycs. B 1 pht 1 ihv. A 1 vie 1 1 1 abo. A 1 ycs. B 1 pht 1 ihv. A 1 vie 36 39 51 27 28 . NLFVALYDfvasgdntlsitk. GEKLRVLgynhn. . . g. E k. GVIYALWDyepqnddelpmke. GDCMTIIhrede. . . dei. E g. YQYRALYDykkereedidlhl. GDILTVNkgslvalgfsdgqearpeei. G. NFRVYYRDsrd. . . pvwk. GPAKLLWkg. . . . e. G. drvrkksga. . awq. GQIVGWYctnlt. . . pe. G WCEAQt. . kngq. GWVPSNYITPVN. . . WWWARl. . ndke. GYVPRNLLGLYP. . . WLNGYnettger. GDFPGTYVEYIGrkkisp AVVIQd. . nsdi. KVVPRRKAKIIRd. . . YAVESeahpgsv. QIYPVAALERIN. . . Key alpha helix RED beta strand GREEN core blocks UNDERSCORE BAli. BASE Reference alignments
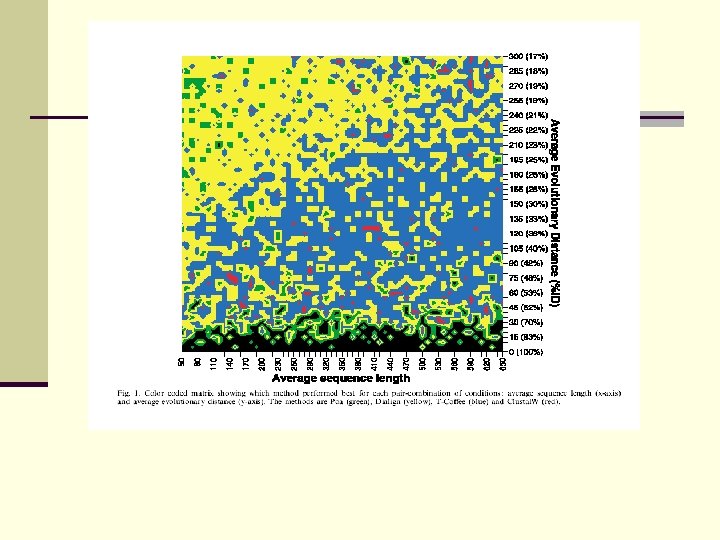

Result: DIALIGN best method for distantly related sequences, T-Coffee best for globally related proteins

Evaluation of multi-alignment methods BAli. BASE: 5 categories of benchmark sequences (globally related, internal gaps, end gaps) CLUSTAL W, T-COFFEE, MAFFT, PROBCONS perform well on globally related sequences, DIALIGN superior for local similarities

Evaluation of multi-alignment methods Conclusion: no single best multi alignment program! Advice: try different methods!

Anchored sequence alignment Idea: semi-automatic alignment n use expert knowledge to define constraints instead of fully automated alignment n Define parts of the sequences where biologically correct alignment is known as anchor points, align rest of the sequences automatically.

Anchored sequence alignment NLFVALYDFVASGDNTLSITKGEKLRVLGYNHN IIHREDKGVIYALWDYEPQNDDELPMKEGDCMT GYQYRALYDYKKEREEDIDLHLGDILTVNKGSLVALGFS

Anchored sequence alignment NLFVALYDFVASGDNTLSITKGEKLRVLGYNHN IIHREDKGVIYALWDYEPQNDDELPMKEGDCMT GYQYRALYDYKKEREEDIDLHLGDILTVNKGSLVALGFS Anchor points in multiple alignment

Anchored sequence alignment NLFV ALYDFVASGDNTLSITKGEKLRVLGYNHN IIHREDKGVIYALWDYEPQND DELPMKEGDCMT GYQYRALYDYKKEREEDIDLHLGDILTVNKGSLVALGFS Anchor points in multiple alignment

Anchored sequence alignment -------NLF V-ALYDFVAS GD---- NTLSITKGEk lrv. LGYNhn iihredk. GVI Y-ALWDYEPQ ND---- DELPMKEGDC MT-------GYQ Yr. ALYDYKKE REedidlhlg DILTVNKGSL VA-LGFS-- Anchored multiple alignment
 under costraints given")
Algorithmic questions Goal: n Find optimal alignment (=consistent set of fragments) under costraints given by userspecified anchor points!

Algorithmic questions Additional input file with anchor points: 1 2 1 3 3 4 215 34 317 231 78 402 5 23 8 4. 5 1. 23 8. 5

Algorithmic questions NLFVALYDFVASGDNTLSITKGEKLRVLGYNHN IIHREDKGVIYALWDYEPQNDDELPMKEGDCMT GYQYRALYDYKKEREEDIDLHLGDILTVNKGSLVALGFS

Algorithmic questions Additional input file with anchor points: 1 2 1 3 3 4 215 34 317 231 78 402 5 23 8 4. 5 1. 23 8. 5

Algorithmic questions Additional input file with anchor points: 1 2 1 3 3 4 Sequences 215 34 317 231 78 402 5 23 8 4. 5 1. 23 8. 5

Algorithmic questions Additional input file with anchor points: 1 2 1 3 3 4 Sequences 215 34 317 231 78 402 start positions 5 23 8 4. 5 1. 23 8. 5

Algorithmic questions Additional input file with anchor points: 1 2 1 3 3 4 Sequences 215 34 317 231 78 402 start positions 5 23 8 length 4. 5 1. 23 8. 5

Algorithmic questions Additional input file with anchor points: 1 2 1 3 3 4 Sequences 215 34 317 231 78 402 start positions 5 23 8 4. 5 1. 23 8. 5 length score
- Slides: 89