Bioinformatics and Proteomics March 28 2003 NIH Proteomics

Bioinformatics and Proteomics March 28, 2003 NIH Proteomics Workshop Bethesda, MD Anastasia Nikolskaya, Ph. D. Research Assistant Professor Protein Information Resource Department of Biochemistry and Molecular Biology Georgetown University Medical Center

Overview • Role of Bioinformatics/Computational Biology in Proteomics Research • Genomics • Functional Annotation of Proteins • Classification of Proteins • Bioinformatics Databases and Analytical Tools (Dr. Yeh and Dr. Hu) Sequence function

Functional Genomics/Proteomics studies biological systems based on global knowledge of genomes, transcriptomes, proteomes, metabolomes. Functional genomics studies biological functions of proteins, complexes, pathways based on the analysis of genome sequences. Includes functional assignments for protein sequences. – Genome: All the Genetic Material in the Chromosomes – Transcriptome: Entire Set of Gene Transcripts – Proteome: Entire Set of Proteins – Metabolome: Entire Set of Metabolites Genome Transcriptome Proteome Metabolome

Proteomics • Data: Gene Expression Profiling - Genome-Wide Analyses of Gene Expression • Data: Structural Genomics - Determine 3 D Structures of All Protein Families • Data: Genome Projects (Sequencing) - Functional genomics - Knowing complete genome sequences of a number of organisms is the basis of the proteomics research

DNA Sequence Genomic DNA Sequence Gene Recognition Gene Protein Sequence Func tion Promoter C A C A A T Exon 1 5' UTR T A A T Intron G T Exon 2 A G Exon 3 Intron G T 3' UTR A A T A A A G G A Protein Sequence Exon 1 Structure Determination Family Classification Protein Structure Exon 2 Exon 3 Function Analysis Protein Family Molecular Evolution Gene Network Metabolic Pathway

Bioinformatics and Genomics/Proteomics Sequence, Other Data Unknown Genes Pathways and Regulatory Circuits Putative Functional Groups Hypothetical Cell

Most new proteins come from genome sequencing projects • • • Mycoplasma genitalium - 484 proteins Escherichia coli - 4, 288 proteins S. cerevisiae (yeast) - 5, 932 proteins C. elegans (worm) ~ 19, 000 proteins Homo sapiens ~ 40, 000 proteins. . . and have unknown functions

Advantages of knowing the complete genome sequence • All encoded proteins can be predicted and identified • The missing functions can be identified analyzed • Peculiarities and novelties in each organism can be studied • Predictions can be made and verified

The changing face of protein science 20 th century 21 st century • Few well-studied • Many “hypotheti proteins cal” proteins • Mostly globular • Various, often with enzymatic no enzymatic activity • Biased protein • Natural protein set

Properties of the natural protein set • Unexpected diversity of even common enzymes (analogous, paralogous, xenologous, etc. enzymes ) • Conservation of the reaction chemistry, but not the substrate specificity • Functional diversity in closely related proteins • Abundance of new structures

Objectives of functional analysis for different groups of proteins • Experimentally characterized Best annotated protein database: Swiss. Prot • “Knowns” = Characterized by similarity (closely related to experimentally characterized) – Make sure the assignment is plausible • Function can be predicted – Extract maximum possible information – Avoid errors and overpredictions – Fill the gaps in metabolic pathways • “Unknowns” (conserved or unique) – Rank by importance

E. coli M. jannaschii S. cerevisiae H. sapiens Characterized experimentally 2046 97 3307 10189 Characterized by similarity 1083 1025 1055 10901 Unknown, conserved 285 211 1007 2723 Unknown, no similarity 874 411 966 7965 from Koonin and Galperin, 2003, with modifications

Problems in functional assignments for “knowns” • Previous low quality annotations - misinterpreted experimental results (e. g. suppressors, cofactors) - biologically senseless annotations Deinococcus: head morphogenesis protein Arabidopsis: separation anxiety protein-like Helicobacter: brute force protein Methanococcus: centromere-binding protein Plasmodium: frameshift - propagated mistakes of sequence comparison

Problems in functional assignments for “knowns” • Multi-domain organization of proteins Histidine kinase Periplasmic sensor domain His kinase domain Uncharacterized domain
 •")
Problems in functional assignments for “knowns” • Low sequence complexity (coiled-coil, non-globular regions) • Non-orthologous gene displacement • Enzyme evolution (divergence in sequence and function)

Enzyme recruitment: Minor mutational changes convert a glycerol kinase into gluconate kinase Differences between gluconate and glycerol/xylulose kinases Leads to non-orthologous gene displacement

Objectives of functional analysis for different groups of proteins • Experimentally characterized • “Knowns” = Characterized by similarity (closely related to experimentally characterized) – Make sure the assignment is plausible • Function can be predicted – Extract maximum possible information – Avoid errors and overpredictions – Fill the gaps in metabolic pathways • “Unknowns” (conserved or unique) – Rank by importance

Functional Prediction: Dealing with “hypothetical” proteins • Computational analysis – Sequence analysis of the new ORFs • Mutational analysis • Functional analysis – Expression profiling – Tracking of cellular localization • Structural analysis – Determination of the 3 D structure

Structural Genomics Protein Structure Initiative: Determine 3 D Structures of All Proteins – Family Classification: Organize Protein Sequences into Families, collect families without known structures – Target Selection: Select Family Representatives as Targets – Structure Determination: X-Ray Crystallography or NMR Spectroscopy – Homology Modeling: Build Models for Other Proteins by Homology – Functional prediction based on structure
 Contains bound ATP")
Structural Genomics: Structure-Based Functional Assignments Methanococcus jannaschii MJ 0577 (Hypothetical Protein) Contains bound ATP => ATPase or ATP-Mediated Molecular Switch Confirmed by biochemical experiments

Crystal structure is not a function!
 • Detailed manual analysis of sequence similarities")
Improving functional assignments for “unknowns” (Functional Prediction) • Detailed manual analysis of sequence similarities • Cluster analysis of protein families (family databases) • Use of sophisticated database searches (PSI-BLAST, HMM)

Using comparative genomics for protein analysis • Those amino acids that are conserved in divergent proteins (archaeal and bacterial, hyperthermophilic and mesophilic) are likely to be important for catalytic activity. • Comparative analysis allows us to find subtle sequence similarities in proteins that would not have been noticed otherwise • Prediction of the 3 D fold and general function is much easier than prediction of exact biological (or biochemical) function.

Using comparative genomics for protein analysis • For some reason, the reaction chemistry often remains conserved even when sequence diverges almost beyond recognition • Sequence database searches that use exotic or highly divergent query sequences often reveal more subtle relationships than those using queries from humans or standard model organisms (E. coli, yeast, worm, fly). • Sequence analysis complements structural comparisons and can greatly benefit from them

Poorly characterized protein families • Enzyme activity can be predicted, the substrate remains unknown (ATPases, GTPases, oxidoreductases, methyltransferases, acetyltransferases) • Helix-turn-helix motif proteins (predicted transcriptional regulators) • Membrane transporters

Improving functional assignments for “unknowns” • Phylogenetic distribution – Wide - most likely essential – Narrow - probably clade-specific – Patchy - most intriguing, niche-specific • Domain association – Rosetta Stone for multidomain proteins • Gene neighborhood (operon organization)

Using genome context for functional prediction
")
Problems in functional assignments/predictions • Identification of protein-coding regions • Delineation of potential function(s) for distant paralogs • Identification of domains in the absense of close homologs • Analysis of proteins with low sequence complexity

“Unknown unknowns” • Phylogenetic distribution – Wide - most likely essential – Narrow - probably clade-specific – Patchy - most intriguing, nichespecific

To deal with the ocean of new sequences, need “natural” protein classification Discovery of New Knowledge by Using Information Embedded within Families of Homologous Sequences and Their Structures • Protein families are real and reflect evolutionary relationships • Protein classification systems can be used to – Improve sensitivity of protein identification – Provide new protein sequence annotation, simplifying the search for non-obvious relationships – Detect and correct genome annotation errors systematically – Drive other annotations (actve site etc) – Provide basis for evolution, genomics and proteomics research

The ideal system would be: • Comprehensive, with each sequence classified either as a member of a family or as an “orphan” sequence, a family of one • Hierarchical, with families united into superfamilies on the basis of distant homology • Allow for simultaneous use of the whole protein and domain information (domains mapped onto proteins) • Allow for automatic classification/annotation of new sequences when these sequences are classifiable into the existing families • Expertly curated (family name, function, evidence attribution (experimental vs predicted), background etc). This is the only way to avoid annotation errors and prevent error propagation

The ideal system has yet to be created, but there are several very useful systems

Levels of Protein Classification Class / Composition of structural elements No relationships Fold TIM-Barrel Topology of folded backbone Possible monophyly above and below Superfamily Aldolase Recognizable sequence similarity (motifs); basic biochemistry Monophyletic origin Family Class I Aldolase High sequence similarity (alignments); biochemical properties Evolution by ancient duplications COG 2 -keto-3 -deoxy-6 - Orthology for a given set of Origin traceable to phosphogluconate species; biochemical a single gene in aldolase activity; biological function LCA LSE PA 3131 and PA 3181 Paralogy within a lineage Evolution by recent duplication and loss
 •")
Protein Evolution • Tree of Life & Evolution of Protein Families (Dayhoff, 1978) • Can build a tree representing evolution of a protein family, based on sequences • Othologus Gene Family: Organismal and Sequence Trees Match Well

Protein Evolution • • • Homolog – Common Ancestors – Common 3 D Structure – Common Active Sites or Binding Domains Ortholog – Derived from Speciation Paralog – Derived from Duplication

M T Craniata a i b i h p m A i m sto o e l e e a d i n yxi t e T r t a r b tr e Ve a Myo (Rat) Hb. A (Rat) Hb. B (Rat) Myo (Frog) Hb. A (Frog) Hb. B (Frog) Myo (Cod) Hb. A (Cod) Hb. B (Cod) Hb (Hagfish) Myo (Hagfish) Orthologs and Paralogs M am m a d o p a ia l a
 Hb. A (Rat) Hb. B (Rat) Myo")
COG myoglobins LCA of Craniata Myo (Rat) Hb. A (Rat) Hb. B (Rat) Myo (Frog) Hb. A (Frog) Hb. B (Frog) Myo (Cod) Hb. A (Cod) Hb. B (Cod) Hb (Hagfish) Myo (Hagfish) Orthologs and Paralogs COG hemoglobins
 Orthologs (COG Myo) Myo (Cod) Myo (Frog) Myo (Rat)")
Orthologs and Paralogs Myo (Hagfish) Orthologs (COG Myo) Myo (Cod) Myo (Frog) Myo (Rat) Out-paralogs (globin family) Orthologs (COG Hb) Hb (Hagfish) Hb. A (Cod) Hb. A (Frog) Hb. A (Rat) Hb. B (Cod) Hb. B (Frog) Hb. B (Rat) Sub. COG In-paralogs (LSE in Vertebrata) Sub. COG
 Hb. A (Cod) Hb. B (Cod) Myo (Frog) Hb. A (Frog) Hb.")
Myo (Cod) Hb. A (Cod) Hb. B (Cod) Myo (Frog) Hb. A (Frog) Hb. B (Frog) Myo (Rat) Hb. A (Rat) Hb. B (Rat) Hb (Hagfish) Myo (Hagfish) Orthologs and Paralogs COG myoglobins COG hemoglobins A COG hemoglobins LCA of Vertebrata
 Out-paralogs (globin family) Orthologs (COG Hb. A) Orthologs")
Orthologs and Paralogs Orthologs (COG Myo) Out-paralogs (globin family) Orthologs (COG Hb. A) Orthologs (COG Hb. B) Myo (Cod) Myo (Frog) Myo (Rat) Hb. A (Cod) Hb. A (Frog) Hb. A (Rat) Hb. B (Cod) Hb. B (Frog) Hb. B (Rat)

Levels of Protein Classification Class / Composition of structural elements No relationships Fold TIM-Barrel Topology of folded backbone Possible monophyly above and below Superfamily Aldolase Recognizable sequence similarity (motifs); basic biochemistry Monophyletic origin Family Class I Aldolase High sequence similarity (alignments); biochemical properties Evolution by ancient duplications COG 2 -keto-3 -deoxy-6 - Orthology for a given set of Origin traceable to phosphogluconate species; biochemical a single gene in aldolase activity; biological function LCA LSE PA 3131 and PA 3181 Paralogy within a lineage Evolution by recent duplication and loss

Protein Family-Domain-Motif • Domain: Evolutionary/Functional/Structural Unit Domain = structurally compact, independently folding unit that forms a stable three-dimentional structure and shows a certain level of evolutionary conservation. Usually, corresponds to an evolutionary unit. A protein can consist of a single domain or multiple domains. Proteins have modular structure. • Motif: Conserved Functional/Structural Site

Protein Evolution: Sequence Change vs. Domain Shuffling If enough similarity remains, one can trace the path to the common origin What about these?
 SF 001501 CM (Aro. Q type) PDH")
Recent Domain Shuffling CM (Aro. Q type) SF 001501 CM (Aro. Q type) PDH PDH CM (Aro. Q type) SF 006786 SF 001499 ACT SF 005547 PDT ACT SF 001424 PDT ACT SF 001500

Protein classification: proteins and domains • Option 1: classify domains - take individual domain sequences, consider them as independently evolving units and build a classification system - allows to go all the way to the deepest possible level, the last point of traceable homology and common origin (fold) - domain databases (Pfam, SMART, CDD) allow to map domains onto a query sequence

Protein classification: proteins and domains • Option 2: classify full-length proteins - In cases of multidomain proteins, does not allow to go deep along the evolutionary tree - All proteins in a family will often have a common biological function, which is very convenient for annotation - Domains will be mapped onto protein families

Practical Classification of Proteins: Setting Realistic Goals We strive to reconstruct the natural classification of proteins to the fullest possible extent BUT Domain shuffling rapidly degrades the continuity in the protein structure (faster than sequence divergence degrades similarity) THUS The further we extend the classification, the finer is the domain structure we need to consider SO We need to compromise between the depth of analysis and protein integrity

Clasification: current status • PIR Superfamilies: Proteins in PIRPSD: 283, 289 Proteins classified: 187, 871 2/3 of the PIR proteins • COGs: ~ 70% of each microbial genome ~ 50% of each Eukaryotic genome in 3 -clade COG ~ 20% ? of each Eukaryotic genome in LSEs
")
PIR Web Site (http: //pir. georgetown. edu)
 Proteins – Homeomorphic (Common Domain Architecture) – Monophyletic")
PIR Superfamily Concept – Whole (Full-Length) Proteins – Homeomorphic (Common Domain Architecture) – Monophyletic (Common Evolutionary Origin) – Hierarchical structure (Family and Superfamily) – Non-Overlapping placement within each level PIR Superfamily vs. Other Concepts – Evolution: Superfamily hierarchy reflects orthology and paralogy – Structure: PIR superfamily generally corresponds to SCOP family – Domain: Domains are mapped onto the Superfamily – Motif: Motifs (functional/structural sites) are mapped onto the Superfamily – Function: a Superfamily may contain divergent functions

PIR Superfamilies • Created by automated clustering by % identity with coverageby-length requirements. Creation of new Superfamilies is an ongoing process. • Automated classification rules are refined by expert curation: - Evolution rates are very different in different “branches” of the protein universe, so need very different score cutoffs - Verify/add members • Annotation (at level of orthology): Superfamily Name, Description, Bibliography • In some cases, more than one orthologous group will be included into a single Superfamily; these Superfamilies will often be very large and diverse • Depth of hierarchy will be different for single-domain and multidomain proteins This is work in progress and will become available through PIR (i. Pro. Class) and Inter. Pro
, Aro. Q class – – SF 001501 –")
CM-Related Superfamilies • Chorismate Mutase (CM), Aro. Q class – – SF 001501 – CM (Prokaryotic type) [PF 01817] SF 001499 – tyr. A bifunctional enzyme (Prok) [PF 01817 -PF 02153] SF 001500 – phe. A bifunctional enzyme (Prok) [PF 01817 -PF 00800] SF 017318 – CM (Eukaryotic type) [Regulatory Dom-PF 01817] • Chorismate Mutase, Aro. H class – SF 005965 – CM [PF 01817] Aro. Q Prok Aro. Q Euk Aro. H
")
i. Pro. Class Superfamily Report (I)
")
i. Pro. Class Superfamily Report (II)

Inter. Pro -Inter. Pro is an integrated resource for protein families, domains and sites. - Inter. Pro combines a number of databases that use different methodologies. By uniting the member databases, Inter. Pro capitalizes on their individual strengths, producing a powerful integrated diagnostic tool. Member databases: PROSITE, PRINTS, Pfam, SMART, Pro. Dom, and TIGRFAMs PIR to be added soon SWISSPROT and Tr. EMBL matches used as examples

Inter. Pro Entry Type defines the entry as a Family, Domain, Repeat, or Posttranslational modification site (other sites to be added: binding site, active site). Family = protein family. PIR SFs will generally belong to this type. “Contains” field lists domains within this protein “Found in”: for domain entries, lists families which contain this domain

PIR Superfamilies are being integrated into Inter. Pro Entry Type = Family SF 001500 Bifunctional chorismate mutase / prephenate dehydratase (P-protein) CM PDT ACT

COGs Clusters of Orthologous Groups -complete genomes - reciprocal best hits - no score cutoffs Comparative genomics - a branch of computational biology that uses complete genome sequences

Construction of COGs: Genome 1 Genome 2

Construction of COGs: Yeast Bidirectional best hit E. coli fbp YLR 377 c Triangle Bidirectional the simplest best hit COG Bidirectional best hit Synechocystis slr 0952

Construction of COGs: Merge triangles

Construction of COGs: Add all homologs New protein ? Yeast YLR 377 c E. coli fbp Synechocystis slr 0952
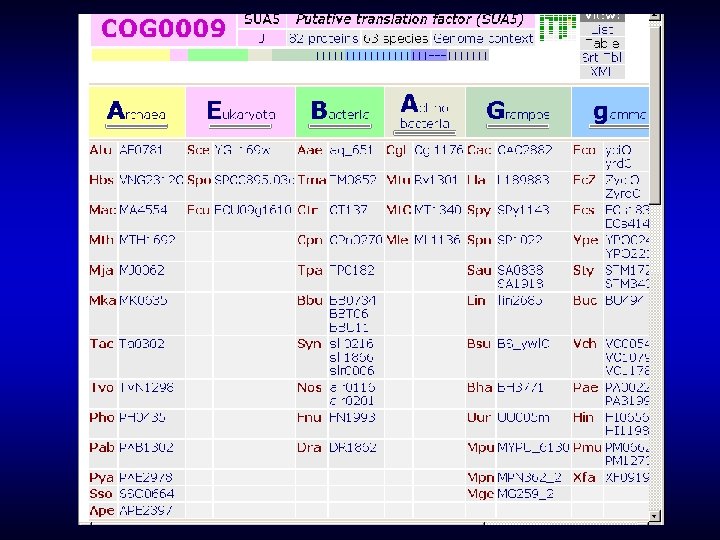
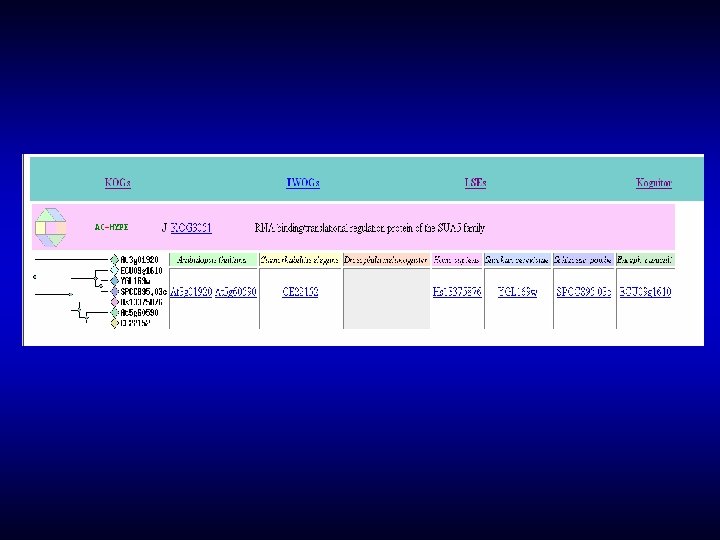
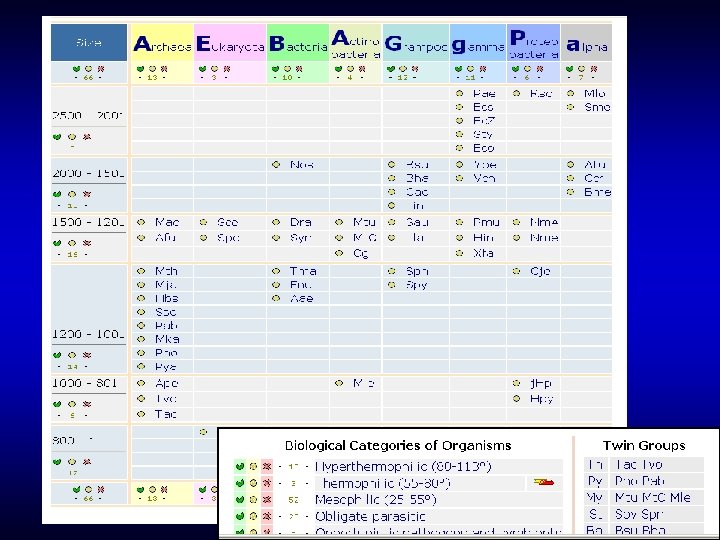

In COGs, the dilemma between the depth of analysis and protein integrity is approached by keeping proteins intact whenever possible, and dividing into modules (single- or multidomain) when necessary

Case Study 1: Prediction verified: GGDEF domain • Proteins containing this domain: Caulobacter crescentus Ple. D controls swarmer cell - stalk cell transition (Hecht and Newton, 1995). In Rhizobium leguminosarum, Acetobacter xylinum, required for cellulose biosynthesis (regulation) • Predicted to be involved in signal transduction because it is found in fusions with other signaling domains (receiver, etc) • In Acetobacter xylinum, cyclic di-GMP is a specific nucleotide regulator of cellulose synthase (signalling molecule). Multidomain protein with GGDEF domain was shown to have diguanylate cyclase activity (Tal et al. , 1998) • Detailed sequence analysis tentatively predicts GGDEF to be a diguanylate cyclase domain (Pei and Grishin, 2001) • Complementation experiments prove diguanylate cyclase activity of GGDEF (Ausmees et al. , 2001)
 Che. Y-like")
Case study 2: Defining a novel domain family Prokaryotic Response Regulatiors (RRs) Che. Y-like receiver Output Variable - DNA-binding - Enzymatic What if domain is not described yet? Che. Y receiver PSY-BLAST with C-terminal portion alone
![Two Groups of Unusual RRs [Receiver-X] SF 006198, COG 3279 1. Alg. R-related •](http://slidetodoc.com/presentation_image/10367fcf4f60fbb2b472adf3f02d0e29/image-69.jpg "Two Groups of Unusual RRs [Receiver-X] SF 006198, COG 3279 1. Alg. R-related •")
Two Groups of Unusual RRs [Receiver-X] SF 006198, COG 3279 1. Alg. R-related • Pseudomonas aeruginosa (Alg. R): alginate biosynthesis • Klebsiella pneumoniae (Mrk. E): formation of adhesive fimbriae • Clostridium perfringens (Vir. R): virulence factors 2. Regulators of autoinduced peptide-controlled regulons • Staphylococcus aureus (Agr. A): virulence factors • Lactobacillus plantarum (Pln. C, Pln. D): bacteriocin production • Streptococcus pneumoniae (Com. E): competence Properties of the Che. Y- Lyt. TR transcriptional regulators • • Regulate secreted and extracellular factors Often regulate their own expression Bind to imperfect direct repeat sites in -80 to - 40 area (or in UAS) Can be phosphorylated by His kinases, but form operons with His. K-type sensor ATPases • Contain a conserved Lyt. TR-type DNA-binding domain

Lyt. TR - a new DNA-binding domain not similar to HTH, winged helix, or ribbon-helix DNA-binding domains

Domain organization of Lyt. TR proteins other than Che. Y-Lyt. TR Stand-alone Lyt. TR Streptococcus pneumoniae Blp. S Pseudomonas phage D 3 Orf 50 40 aa - Lyt. TR Lactococcus lactis L 121252 Listeria monocytogenes Lmo 0984 Staphylococcus aureus SA 2153 Streptococcus pneumoniae SP 0161 ABC - Lyt. TR Bacillus halodurans BH 3894 MHYT - Lyt. TR Oligotropha carboxydovora Cox. C, Cox. H 3 TM - Lyt. TR Xanthomonas campestris Rpf. D Caulobacter crescentus CC 1610 Mesorhizobium loti mll 0891 Caulobacter crescentus CC 0295 Caulobacter crescentus CC 0330, CC 3036 Caulobacter crescentus CC 0551 3 TM - Lyt. TR 4 TM - Lyt. TR 8 TM - Lyt. TR PAS - Lyt. TR Burkholderia cepacia Geobacter sulfurreducens

Consensus binding site for the Lyt. TR domains

Predicted Lyt. TR-regulated genes Expected Bacillus subtilis Oligotropha carboxidovorans Staphylococcus aureus Streptococcus pneumoniae Unexpected Bacillus subtilis Staphylococcus aureus nat. AB (Na+-ATPase) com. C, com. H (CO growth) lrg. AB (autolysis) hld (hemolysin delta) alr, din. B, rap. I, veg, yba. J, ybb. I, yce. A, ydb. S, ydj. L, yeb. B, yfi. V, yku. A cap. O, coa, hsd. R, SA 0096, SA 0257, SA 0285, SA 0302, SA 0357, SA 0358, SA 0513

Impact of genomics • Single protein level – Discovery of new enzymes and superfamilies – Prediction of active sites and 3 D structures • Pathway level – Identification of “missing” enzymes – Prediction of alternative enzyme forms – Identification of potential drug targets • Cellular metabolism level – Multisubunit protein systems – Membrane energy transducers – Cellular signaling systems

Examples for analysis: 1. Retrieve one of the following protein sequences: PIR: C 69086 D 64376 Gen. Bank GI: 15679635. Using analysis tools available on the web, check if the functional annotation is correct, and provide correct annotation without looking at internal PIR or COG annotations (Run BLAST with CDsearch and SMART to start with). When you are done, look at the PIR curated SF annotation (still at internal site only): http: //pir. georgetown. edu/test-cgi/sf/pirclassif. pl? id=SF 006549 http: //pir. georgetown. edu/cgi-bin/ipc. SF 1? id=SF 006549 (compare with original automatic SF annotation at the public site), and at COG annotations. What caused the wrong annotations? In BLAST outputs for these sequences, do you see other wrongly annotated proteins? • Next, analyze the C-terminal domain of these proteins by PSI-BLAST (and alignment analysis) and suggest any speculations as to its function (homework).

Examples for analysis: 2. • Retrieve the following sequence: GI: 7019521 • Take a look at the associated publication (reference). • Analyze the sequence to see if any additional information can be obtained (run PSI-BLAST, and (as a homework) construct multiple alignment). • Take a look at taxonomy report: what does it tell you? • Find experimental paper associated with one of the sequences found by PSI-BLAST. What annotation is appropriate for this sequence and for the entire family?

Examples for analysis: 3. Predict the function of the following proteins: • Gen. Bank: GI: 27716853 • E. coli Yje. E protein Verify and/or correct the following functional annotations. Can you explain why the erroneous annotations were made? • PIR: H 87387 • Gen. Bank: GI: 15606003 GI: 15807219 • PIR: F 70338

Examples for analysis: 4. Homework: an exercise in transitive relationships: Start with >gi|20093648|ref|NP_613495. 1| Uncharacterized membrane protein, conserved in Archaea [Methanopyrus kandleri AV 19] (this is a short membrane protein); run PSI-BLAST, make sure you have filtering, complexity and CD-search off. There are no good hits but a bunch of sub-threshold ones. Collect "suspect" relations, use them as queries and expand the net. You will be able to come up with two proteins: >gi|21227474|ref|NP_633396. 1| hypothetical protein [Methanosarcina mazei Goe 1] and >gi|14324537|dbj|BAB 59464. 1| hypothetical protein [Thermoplasma volcanium] When used as a PSI-BLAST query, the first will tie the Methanopyrus protein into a group, while the second will tie this group to the Sec 61 subunit of preprotein translocase. Then, of course, you can obtain the same result with CD-search in a single step .
- Slides: 78