ASUHAN KEPERAWATAN PASIEN DENGAN THALASEMIA Ns M Shodikin








 Thalasemia")
")









.")




 Selama Masa Kehamilan, hendaknya perlu dikaji")
 Keadaan umum")
 Mata dan konjungtiva terlihat pucat kekuningan 4) Mulut dan bibir terlihat pucat kehitaman")
 Pertumbuhan organ seks sekunder untuk anak pada usia pubertas Ada keterlambatan kematangan seksual,")
 Transpusi")
 Pemberian Roborantia, hindari preparat yang mengandung zat besi. 4) Pemberian Desferioxamin untuk menghambat")

")
 berhubungan dengan hepatomegali/splenomegali yang diatandai dengan")

- Slides: 37
ASUHAN KEPERAWATAN PASIEN DENGAN THALASEMIA Ns. M. Shodikin, M. Kep, Sp, Kep, MB
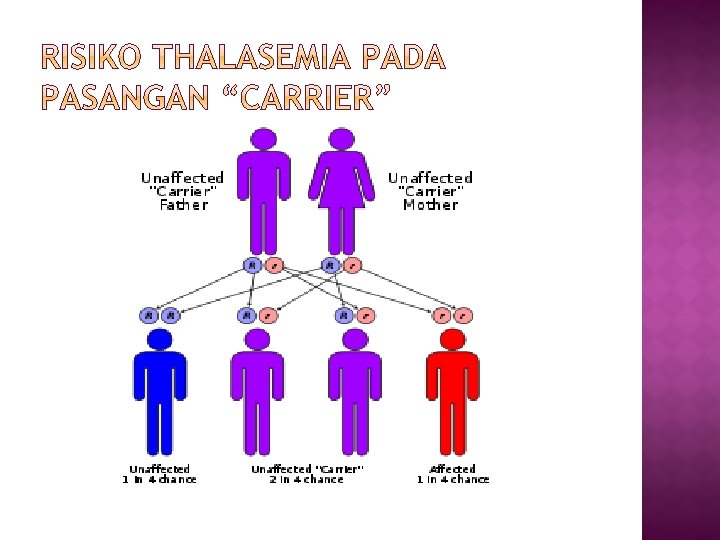
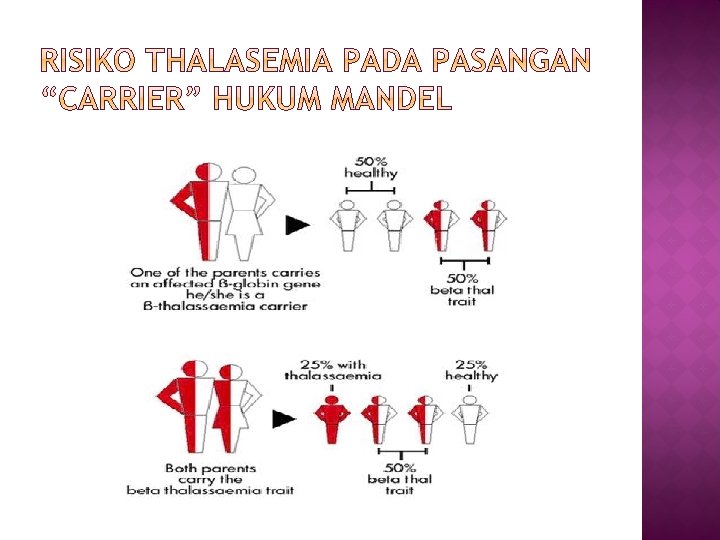
� Talasemia merupakan salah satu jenis anemia hemolitik dan merupakan heriditer yang diturunkan secara autosomal yang paling banyak dijumpai di Indonesia 6 – 10 dari setiap 100 orang Indonesia membawa gen penyakit ini. � Kalau pasangan carrier menikah, kemungkinan untuk mempunyai anak penderita talasemia berat adalah 25%, 50% menjadi pembawa sifat (carrier) talasemia, dan 25% kemungkinan bebas talasemia. � Sebagian besar penderita talasemia adalah anak usia 0 hingga 18 tahun.
� Talasemia adalah sekelompok heterogen gangguan genetic pada sintesis Hb yang ditandai dengan tidak ada atau berkurangnya sintesis rantai globin. � Pada thalasemia alfa, sintesis rantai alfa globin berkurang. � pada thalasemia beta, sintesis rantai beta globin tidak ada (di beri nama thalasemia β 0) atau sangat berkurang (thalasemia β+). � thalasemia terjadi akibat kelainan kuantitatif sintesis rantai globin. Sumber: http: //id. shvoong. com/medicine-andhealth/1958434 -definisithalasemia/#ixzz 1 x 5 GWXOX 6
� Pada talasemia terjadi kelainan pada gen-gen yang mengatur pembentukan dari rantai globin sehingga produksinya terganggu. � Gangguan dari pembentukan rantai globin ini akan mengakibatkan kerusakan pada sel darah merah yang pada akhirnya akan menimbulkan pecahnya sel darah tersebut. Berdasarkan dasar klasifikasi tersebut, maka terdapat beberapa jenis talasemia, yaitu talasemia alfa, beta, dan delta.
� Pada talasemia alfa, terjadi penurunan sintesis dari rantai alfa globulin. Dan kelainan ini berkaitan dengan delesi pada kromosom 16. Akibat dari kurangnya sintesis rantai alfa, maka akan banyak terdapat rantai beta dan gamma yang tidak berpasangan dengan rantai alfa. Maka dapat terbentuk tetramer dari rantai beta yang disebut Hb. H dan tetramer dari rantai gamma yang disebut Hb Barts. Talasemia alfa sendiri memiliki beberapa jenis.
Delesi pada empat rantai alfa � Dikenal juga sebagai hydrops fetalis. Biasanya terdapat banyak Hb Barts. Gejalanya dapat berupa ikterus, pembesaran hepar dan limpa, dan janin yang sangat anemis. Biasanya, bayi yang mengalami kelainan ini akan mati beberapa jam setelah kelahirannya atau dapat juga janin mati dalam kandungan pada minggu ke 36 -40. Bila dilakukan pemeriksaan seperti dengan elektroforesis didapatkan kadar Hb adalah 80 -90% Hb Barts, tidak ada Hb. A maupun Hb. F. Delesi pada tiga rantai alfa � Dikenal juga sebagai Hb. H disease biasa disertai dengan anemia hipokromik mikrositer. Dengan banyak terbentuk Hb. H, maka Hb. H dapat mengalami presipitasi dalam eritrosit sehingga dengan mudah eritrosit dapat dihancurkan. Jika dilakukan pemeriksaan mikroskopis dapat dijumpai adanya Heinz Bodies.
Delesi pada dua rantai alfa � Juga dijumpai adanya anemia hipokromik mikrositer yang ringan. Terjadi penurunan dari Hb. A 2 dan peningkatan dari Hb. H. Delesi pada satu rantai alfa � Disebut sebagai silent carrier karena tiga lokus globin yang ada masih bisa menjalankan fungsi normal.
� Disebabkan karena penurunan sintesis rantai beta. Dapat dibagi berdasarkan tingkat keparahannya, yaitu talasemia mayor, intermedia, dan karier. Pada kasus talasemia mayor Hb sama sekali tidak diproduksi. Mungkin saja pada awal kelahirannya, anak-anak talasemia mayor tampak normal tetapi penderita akan mengalami anemia berat mulai usia 3 -18 bulan. Jika tidak diobati, bentuk tulang wajah berubah dan warna kulit menjadi hitam. Selama hidupnya penderita akan tergantung pada transfusi darah. Ini dapat berakibat fatal, karena efek sampingan transfusi darah terus menerus yang berupa kelebihan zat besi (Fe)[3]. Salah satu ciri fisik dari penderita talasemia adalah kelainan tulang yang berupa tulang pipi masuk ke dalam dan batang hidung menonjol (disebut gacies cooley), penonjolan dahi dan jarak kedua mata menjadi lebih jauh, serta tulang menjadi lemah dan keropos[4].
� Secara molekuler talasemia dibedakan atas : Thalasemia α (gangguan pembentukan rantai α) Thalasemia ß (gangguan p[embentukan rantai ß) Thalasemia ß-Ð (gangguan pembentukan rantai ß dan Ð yang letak gen nya diduga berdekatan). Thalasemia Ð (gangguan pembentukan rantai Ð)
� Secara klinis talasemia dibagi dalam 2 golongan yaitu : Thalasemia Mayor (bentuk homozigot) Memberikan gejala klinis yang jelas Thalasemia Minor biasanya tidak memberikan gejala klinis
Thalasemia mayor, gejala klinik telah terlihat sejak anak baru berumur kurang dari 1 tahun, yaitu: � Lemah � Pucat � Perkembangan fisik tidak sesuai dengan umur � Berat badan kurang � Tidak dapat hidup tanpa transfusi
Thalasemia intermedia : ditandai oleh anemia mikrositik, bentuk heterozigot. Thalasemia minor/thalasemia trait : � � ditandai oleh splenomegali, anemia berat, bentuk homozigot. Pada anak yang besar sering dijumpai adanya: Gizi buruk � Perut buncit karena pembesaran limpa dan hati yang mudah diraba � Aktivitas tidak aktif karena pembesaran limpa dan hati (Hepatomegali ), Limpa yang besar ini mudah ruptur karena trauma ringan saja
� Gejala khas adalah: Bentuk muka mongoloid yaitu hidung pesek, tanpa pangkal hidung, jarak antara kedua mata lebar dan tulang dahi juga lebar. Keadaan kuning pucat pada kulit, jika sering ditransfusi, kulitnya menjadi kelabu karena penimbunan besi.
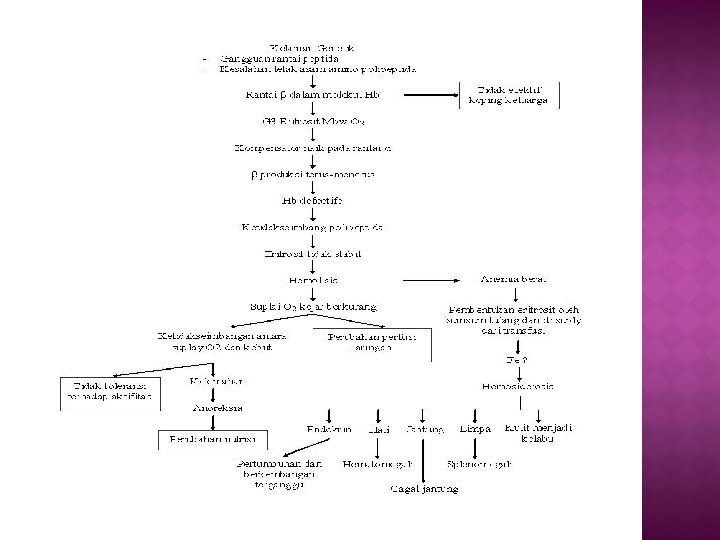
� Penyebab anemia pada thalasemia bersifat primer dan sekunder. Penyebab primer adalah berkurangnya sintesis Hb A dan eritropoesis yang tidak efektif disertai penghancuran selsel eritrosit intrameduler. Penyebab sekunder adalah karena defisiensi asam folat, bertambahnya volume plasma intravaskuler yang mengakibatkan hemodilusi, dan destruksi eritrosit oleh system retikuloendotelial dalam limfa dan hati. � Penelitian biomolekular menunjukkan adanya mutasi DNA pada gen sehingga produksi rantai alfa atau beta dari hemoglobin berkurang. Tejadinya hemosiderosis merupakan hasil kombinasi antara transfusi berulang, peningkatan absorpsi besi dalam usus karena eritropoesis yang tidak efektif, anemia kronis serta proses hemolisis.
Normal hemoglobin adalah terdiri dari Hb-A dengan dua polipeptida rantai alpa dan dua rantai beta. � Pada Beta thalasemia yaitu tidak adanya atau kurangnya rantai Beta dalam molekul hemoglobin yang mana ada gangguan kemampuan eritrosit membawa oksigen. � Ada suatu kompensator yang meninghkatkan dalam rantai alpa, tetapi rantai Beta memproduksi secara terus menerus sehingga menghasilkan hemoglobin defektive. Ketidakseimbangan polipeptida ini memudahkan ketidakstabilan disintegrasi. Hal ini menyebabkan sel darah menjadi hemolisis dan menimbulkan anemia dan atau hemosiderosis. �
Normal hemoglobin adalah terdiri dari Hb-A dengan dua polipeptida rantai alpa dan dua rantai beta. � Pada Beta thalasemia yaitu tidak adanya atau kurangnya rantai Beta dalam molekul hemoglobin yang mana ada gangguan kemampuan eritrosit membawa oksigen. � Ada suatu kompensator yang meninghkatkan dalam rantai alpa, tetapi rantai Beta memproduksi secara terus menerus sehingga menghasilkan hemoglobin defektive. Ketidakseimbangan polipeptida ini memudahkan ketidakstabilan disintegrasi. Hal ini menyebabkan sel darah menjadi hemolisis dan menimbulkan anemia dan atau hemosiderosis. � �
Kelebihan pada rantai alpa pada thalasemia Beta dan Gama ditemukan pada thalasemia alpa. Kelebihan rantai polipeptida ini mengalami presipitasi dalam sel eritrosit. Globin intra-eritrositk yang mengalami presipitasi, yang terjadi sebagai rantai polipeptida alpa dan beta, atau terdiri dari hemoglobin tak stabil -badan Heinz, merusak sampul eritrosit dan menyebabkan hemolisis. � Reduksi dalam hemoglobin menstimulasi bone marrow memproduksi RBC yang lebih. Dalam stimulasi yang konstan pada bone marrow, produksi RBC diluar menjadi eritropoitik aktif. Kompensator produksi RBC terus menerus pada suatu dasar kronik, dan dengan cepatnya destruksi RBC, menimbulkan tidak adekuatnya sirkulasi hemoglobin. Kelebihan produksi dan distruksi RBC menyebabkan bone marrow menjadi tipis dan mudah pecah atau rapuh. �
� Hasil apusan darah tepi didapatkan gambaran perubahan-perubahan sel dara merah, yaitu mikrositosis, anisositosis, hipokromi, poikilositosis, kadar besi dalam serum meninggi, eritrosit yang imatur, kadar Hb dan Ht menurun. � Elektroforesis hemoglobin: hemoglobin klien mengandung Hb. F dan A 2 yang tinggi, biasanya lebih dari 30 % kadang ditemukan hemoglobin patologis.
� � � Hingga kini belum ada obat yang tepat untuk menyembuhkan pasienthalasemia. Transfusi darah diberikan jika kadar Hb telah rendah sekali (kurang dari 6 gr%) atau bila anak terlihat lemah dan tidak ada nafsu makan. Pemberian transfusi hingga Hb mencapai 10 g/dl. Komplikasi dari pemberian transfusi darah yang berlebihan akan menyebabkan terjadinya penumpukan zat besi yang disebut hemosiderosis. Hemosiderosis dapat dicegah dengan pemberian Deferoxamine(desferal). Splenektomi dilakukan pada anak yang lebih tua dari 2 tahun sebelum terjadi pembesaran limpa/hemosiderosis, disamping itu diberikan berbagai vitamin tanpa preparat besi.
A. Pengkajian a. Asal keturunan/kewarganegaraan Thalasemia banyak dijumpai pada bangsa disekitar laut tengah (mediterania). Seperti turki, yunani, Cyprus, dll. Di Indonesia sendiri, thalassemia cukup banyak dijumpai pada anak, bahkan merupakan penyakit darah yang paling banyak diderita.
b. Umur Pada thalasemia mayor yang gejala klinisnya jelas, gejala tersebut telah terlihat sejak anak berumur kurang dari 1 tahun. Sedangkan pada thalasemia minor yang gejalanya lebih ringan, biasanya anak baru datang berobat pada umur sekitar 4 – 6 tahun.
c. Riwayat kesehatan anak Anak cenderung mudah terkena infeksi saluran napas bagian atas infeksi lainnya. Hal ini mudah dimengerti karena rendahnya Hb yang berfungsi sebagai alat transport.
d. Pertumbuhan dan perkembangan adanya kecenderungan gangguan terhadap tumbuh kembang sejak anak masih bayi, karena adanya pengaruh hipoksia jaringan yang bersifat kronik. Hal ini terjadi terutama untuk thalassemia mayor. Pertumbuhan fisik anak adalah kecil untuk umurnya dan ada keterlambatan dalam kematangan seksual, seperti tidak ada pertumbuhan rambut pubis dan ketiak. Kecerdasan anak juga dapat mengalami penurunan. Namun pada jenis thalasemia minor sering terlihat pertumbuhan dan perkembangan anak normal.
e. Pola makan Karena adanya anoreksia, anak sering mengalami susah makan, sehingga berat badan anak sangat rendah dan tidak sesuai dengan usianya. f. Pola aktivitas Anak terlihat lemah dan tidak selincah anak usianya. Anak banyak tidur / istirahat, karena bila beraktivitas seperti anak normal mudah merasa lelah.
g. Riwayat ibu saat hamil (Ante Natal Care) Selama Masa Kehamilan, hendaknya perlu dikaji secara mendalam adanya faktor risiko thalassemia. Sering orang tua merasa bahwa dirinya sehat. Apabila diduga faktor resiko, maka ibu perlu diberitahukan mengenai risiko yang mungkin dialami oleh anaknya nanti setelah lahir.
h. Data keadaan fisik anak thalassemia yang sering didapatkan diantaranya adalah: 1) Keadaan umum Anak biasanya terlihat lemah dan kurang bergairah serta tidak selincah anak seusianya yang normal. 2) Kepala dan bentuk muka Anak yang belum/tidak mendapatkan pengobatan mempunyai bentuk khas, yaitu kepala membesar dan bentuk mukanya adalah mongoloid, yaitu hidung pesek tanpa pangkal hidung, jarak kedua mata lebar, dan tulang dahi terlihat lebar.
3) Mata dan konjungtiva terlihat pucat kekuningan 4) Mulut dan bibir terlihat pucat kehitaman 5) Dada Pada inspeksi terlihat bahwa dada sebelah kiri menonjol akibat adanya pembesaran jantung yang disebabkan oleh anemia kronik 6) Perut Kelihatan membuncit dan pada perabaan terdapat pembesaran limpa dan hati ( hepatosplemagali ). 7) Pertumbuhan fisiknya terlalu kecil untuk umurnya dan BB nya kurang dari normal. Ukuran fisik anak terlihat lebih kecil bila dibandingkan dengan anak-anak lain seusianya.
8) Pertumbuhan organ seks sekunder untuk anak pada usia pubertas Ada keterlambatan kematangan seksual, misalnya, tidak adanya pertumbuhan rambut pada ketiak, pubis, atau kumis. Bahkan mungkin anak tidak dapat mencapai tahap adolesense karena adanya anemia kronik. 9) Kulit Warna kulit pucat kekuning- kuningan. Jika anak telah sering mendapat transfusi darah, maka warna kulit menjadi kelabu seperti besi akibat adanya penimbunan zat besi dalam jaringan kulit (hemosiderosis).
1. Perawatan umum : Makanan dengan gizi seimbang 2. Perawatan khusus : 1) Transpusi darah diberikan bila kadar Hb rendah sekali (kurang dari 6 gr%) atau anak terlihat lemah dan tidak ada nafsu makan. 2) Splenektomi. Dilakukan pada anak yang berumur lebih dari 2 tahun dan bila limpa terlalu besar sehingga risiko terjadinya trauma yang berakibat perdarahan cukup besar.
3) Pemberian Roborantia, hindari preparat yang mengandung zat besi. 4) Pemberian Desferioxamin untuk menghambat proses hemosiderosis yaitu membantu ekskresi Fe. Untuk mengurangi absorbsi Fe melalui usus dianjurkan minum teh. 5) Transplantasi sumsum tulang (bone marrow) untuk anak yang sudah berumur diatas 16 tahun. Di Indonesia, hal ini masih sulit dilaksanakan karena biayanya sangat mahal dan sarananya belum memadai.
DIAGNOSA KEPERAWATAN Gangguan perfusi jaringan b/d penurunan oksigenasi ke sel – sel ditadai dengan pasien mengatakan kepala terasa pusing, warna kulit pucat, bibir tampak kering, sclera ikterik , ekstremitas dingin. Tujuan : gangguan perfusi jaringan teratasi dengan kriteria • Tanda vital normal • Ektremitas hangat • Warna kulit tidak pucat • Sclera tidak ikterik • Bibir tidak kering • Hb normal 12 – 16 gr% INTERVENSI KEPERAWATAN 1. Observasi Tanda Vital. 2. Observasi perfusi : Warna Kulit, rabaan, dan sensasi. 3. Tingkat Kesadaran. 4. Atur Posisi Semi Fowler. 5. Pemberian O 2. 6. Kolaborasi Dengan Dokter Pemberian Tranfusi Darah. 7. Cek Hb serial post trafusi darah.
DIAGNOSA KEPERAWATAN INTERVENSI KEPERAWATAN Devisit volume cairan dan elektrolit berhubungan dengan penurunan input (muntah) ditandai dengan in-take kurang, mukosa mulut kering, turgor kulit lambat kembali, produksi urine kurang. Tujuan : deficit volume cairan dan elektrolit teratasi dengan kriteria: • In-take cukup • Mukosa mulut lembab • Turgor kulit baik • produk urine normal 1. Onservasi Intake Output Cairan. 2. Observasi Tanda Vital. 3. Beri pasien minum sedikit demi sedikit. 4. Terapi cairan secara parenteral sesuai dengan hasil kolaborasi. 5. Cek elektrolit secara berkala.
DIAGNOSA KEPERAWATAN INTERVENSI KEPERAWATAN Gangguan rasa nyaman (nyeri) berhubungan dengan hepatomegali/splenomegali yang diatandai dengan nyeri tekan pada daerah abdomen kwadran kiri atas, abdomen hipertimpani, perut distensi, peristaltic usus 10 x/m Tujuan : gannguan rasa nyaman (nyeri ) teratasi dengan kriteria : • Nyeri abdomen hilang/kurang • Abdomen timpani (perkusi) • Perut tidak distensi 1. 2. 3. 4. Kaji sekala nyeri. Distraksi relaksasi. Posisi semi fowler. Kolaborasi pemnerian analgetik.