Antibody Structure Prediction and the Use of Mutagenesis

Antibody Structure Prediction and the Use of Mutagenesis in Docking Arvind Sivasubramanian, Aroop Sircar, Eric Kim & Jeff Gray Johns Hopkins University, Baltimore. Rosetta. Con 2007.

Antibody structure Ab structure schematic Ab PDB structure
 errors frustrate high-resolution docking Docking of m. Ab 225 (Cetuximab) to")
Homology model (WAM) errors frustrate high-resolution docking Docking of m. Ab 225 (Cetuximab) to its antigen Ab crystal structure Ab homology model Better docking energy 0 25 0 Antigen rmsd relative to native (Å) 25
 Light chain sequence: LTQSPSSLSASVGDRVTITCSASSQVNHMFWYQQ")
Antibody Modeling from Sequence m. Ab AQC 2 (anti-VLA 1) Light chain sequence: LTQSPSSLSASVGDRVTITCSASSQVNHMFWYQQ KPGKAPKPWIYLTSYLASGVPSRFSGSGSGTDYTLT ISSLQPEDFATYYCQQWSGNPWTFGQGTKVE Heavy chain sequence: LVESGGGLVQPGGSLRLSCAASGFTFSRYTMSWVR QAPGKGLEWVAVISGGGHTYYLDSVEGRFTISRDN SKNTLYLQMNSLRAEDTAVYYCTRGFGDGGYFDVW GQGTLVT

Antibody Modeling: Scientific Challenges • Easier aspects of the modeling – Conserved framework structure – Canonical CDR loop conformations – Canonical loops erected on beta-barrel framework • Challenges – CDR H 3 conformation (Loop modeling) – VL-VH orientation (Protein-protein docking)

Step 1: Assemble β-Barrel Antibody sequence Select VL and VH frameworks Assemble β-barrel Graft canonical loops for CDR’s L 1 -L 3, H 1 and H 2 H 3 loop modeling Iteration Refine β-barrel orientation using VL - VH docking Homology model

Step 2: Graft canonical loops Antibody sequence Select VL and VH frameworks Assemble β-barrel Graft canonical loops for CDR’s L 1 -L 3, H 1 and H 2 H 3 loop modeling Iteration Refine β-barrel orientation using VL - VH docking Homology model

Step 3: H 3 modeling Antibody sequence Select VL and VH frameworks Assemble β-barrel Graft canonical loops for CDR’s L 1 -L 3, H 1 and H 2 H 3 loop modeling Iteration Refine β-barrel orientation using V L - VH docking Homology model

CDR 3 loop modeling protocol CDR H 3 sequence Low resolution, fragment-based loop library creation High-resolution loop refinement Loop building w. Rosetta /Ab fragments Pre-screened “kink” fragments One cycle of full-atom refinement Shear, small & CCD moves (5 each) CDR H 3 conformation 25 cycles of DFP minimization

Antibody structure database Download crystal structures from PDB based on SACS database 645 structures Employ Filters (Fc regions, L/H dimer’s, Single chain antibodies) 451 structures Retain structures with R < 2. 5Å 279 structures Eliminate structures with redundant CDR’s 167 structures Database Curation : Automatic VL and VH domain classification, elimination of structures with missing residues, broken loops, Chothia re-numbering. http: //www. bioinf. org. uk/abs/sacs/

CDR H 3 length distribution in antibody structure database

Knowledge base: “Kinked” CDR H 3 C-Terminus conformation Kinked N-Ter C-Ter Extended N-Ter C-Ter

Knowledge base: Conserved rotamers VH VL

Homology modeling: Canonical loop RMSD typically < 1. 0Å 50 L 1 40 Native Framework Number Benchmark PDB’s 30 Homology Framework 20 10 0 40 0. 0 -1. 5 -2. 0 -2. 5 >2. 5 L 3 30 Native Framework Homology Framework 20 40 30 Native Framework 10 0 0 1. 0 -1. 5 -2. 0 -2. 5 >2. 5 Homology Framework 20 10 0. 0 -1. 0 H 2 0. 0 -1. 0 RMSD to Native (Å) 1. 0 -1. 5 -2. 0 -2. 5 >2. 5

Native H 3 recovery: Best RMSD in top 10 models

Native H 3 recovery: High, medium and low accuracy predictions 2 B 2 X RMSD = 0. 31Å 1 DBA RMSD = 1. 00 Å 2 CJU RMSD = 1. 56 Å

CDR H 3 Modeling Number of benchmark PDB's Homology modeling: Best RMSD in top 10 models 8 Total PDB’s = 10 6 Loop length 10 Loop length 9 4 Loop length 8 2 0 1. 0 -1. 5 -2. 0 -2. 5 RMSD (Å) >2. 5 Number of benchmark PDB's Light-heavy chain docking 4 Total PDB’s = 10 3 2 1 0 0. 0 -1. 5 -2. 0 RMSD (Å) 2. 0 -2. 5

Use of antibody-derived fragments improves sampling marginally 1000 decoys are created during the “build” stage with Rosetta+Ab Fragments, with or without bias towards choice of Ab fragments 40 30 Ab frag bias No Ab frag bias % decoys with H 3 RMSD < 1. 25 Å 20 1 kb 5_10 1 qbl_8 1 dba_10 2 jel_9 1 kem_8 2 cju_10 2 b 2 x_10 1 clz_10 1 vfa_8 2 adf_9 1 ynt_9 2 fd 6_9 1 jpt_8 1 igt_9 1 k 4 c_9 1 jhl_9 1 fgn_8 0 1 a 6 t_8 10

3 -residue fragments from H 3 loops cluster at the 0. 5 Å level RMSD (Å)

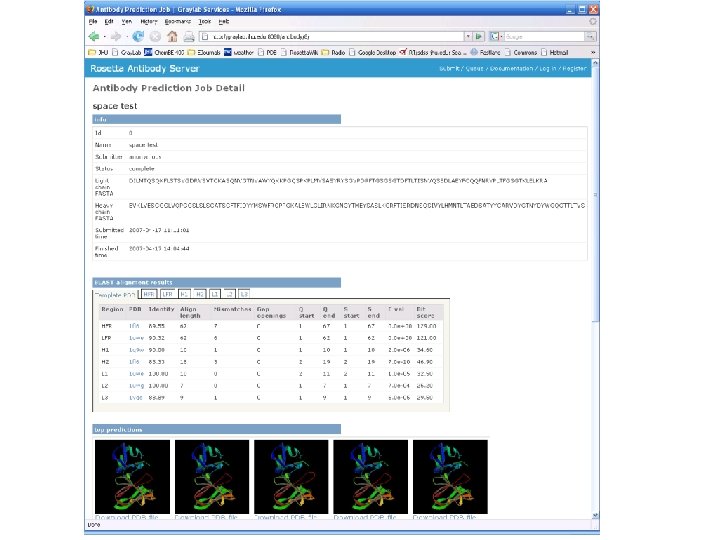
Rosetta Antibody Modeling server • http: //antibody. graylab. jhu. edu/ • Input – Sequences of Light and Heavy Fv chain regions • Outputs – Blast alignments for the framework and CDR loops – Homology model template with all CDR's except H 3

Combing Protein-protein docking and computational mutagenesis Antibody Coordinates Antigen Coordinates Generate docking model using Rosetta. Dock Calculate G of interface mutations in model using Rosetta. Interface No Reject Consistent with experiments? Interface mutations Yes Validate model with new blind computational & experimental mutagenesis

Rosetta. Interface : G values for mutations at protein interfaces Native Complex Mutation Crystal structure or docking model Mutant Complex G = ? ? Interface mutation Rosetta. Interface D 1. 3 – HEL Gcalc Empirical criteria Gexpt 0 < Gcalc < 1. 0 Neutral Gcalc > 1. 0 Binding loss Gcalc Kortemme et al, PNAS, 2002
 Predicted")
Combining Computational & Experimental Mutagenesis (CAPRI Target 21: Orc 1 -Sir 1 complex) Predicted by Aroop Sircar, Arvind Sivasubramanian, Mike Daily No Binding loss 46% of native contacts 1. 92 Å interface rmsd Column 1 is the ΔΔG for our best model Column 2 is the ΔΔG for the native 1 ZHI
 Predicted")
Combining Computational & Experimental Mutagenesis (CAPRI Target 21: Orc 1 -Sir 1 complex) Predicted by Aroop Sircar, Arvind Sivasubramanian, Mike Daily No Binding loss 46% of native contacts 1. 92 Å interface rmsd Column 1 is the ΔΔG for our best model Column 2 is the ΔΔG for the native 1 ZHI The native ORC 1 is more closed than our prediction.

Conclusions • Antibody modeling – Curated antibody structure database – Knowledge-based rules for homology modeling – Canonical loops predicted with 1Å accuracy – H 3 loop accuracy is 1Å in native recovery simulations; homology modeling accuracy is 1 -2Å – Light-heavy chain docking predicted within 1. 5Å accuracy • Mutagenesis and docking – Better discrimination than using simple contact filters

Future work • Antibody modeling – Generate homology models for entire benchmark set – Validate homology models in antibody-antigen docking simulations • Mutagenesis and docking – Implement use of biochemical constraints during docking
- Slides: 27