An Overview of Reporting Unanticipated Problems Involving Risks
 &")
An Overview of Reporting Unanticipated Problems Involving Risks to Subjects or Others (UPIRSOs) & Adverse Events (AEs) to the Yale IRB Human Research Protection Program (HRPP) HRPP COMPLIANCE AND QUALITY TEAM Yale University
 require institutions")
Regulatory Requirements � Federal regulations (45 CFR 46 & 21 CFR 56) require institutions to establish written procedures for ensuring prompt reporting of any unanticipated problems involving risks to subjects or others (UPIRSOs) to the IRB, appropriate institutional officials, and the federal department or agency head. � The FDA has separate regulations that require the prompt reporting of adverse events (or effects) by the investigators to the sponsor and by the sponsor to the FDA as well as to other clinical investigators using the same test article.
 Any incident, experience, or")
Unanticipated Problem Involving Risks to Human Subjects or Others (UPIRSO) Any incident, experience, or outcome that meets ALL 3 of the following criteria: 1. Unexpected (in terms of nature, specificity, severity, or frequency) given (a) the research procedures described in the protocol-related documents, such as the IRB-approved protocol and informed consent document AND (b) the characteristics of the subject population being studied; AND 2. Related or Possibly Related to participation in the research; AND 3. Suggests that the research places subjects or others at greater risk of harm (including physical, psychological, economic, legal, or social harm) than was previously known or recognized.

Adverse Event Any untoward or unfavorable occurrence in a human research subject (physical or psychological harm) temporally associated with the individual’s participation in the research (whether or not considered related to participation in the research).

Serious Adverse Event Any adverse event results in any of the following outcomes: • death; • a life-threatening experience; • inpatient hospitalization or prolongation of existing hospitalization; • a persistent or significant disability/incapacity; • a congenital anomaly/birth defect; OR • based upon appropriate medical judgment, may jeopardize the subject’s health and may require medical or surgical intervention to prevent one of the other outcomes listed in this definition.

Which Adverse Events are considered UPIRSOs?

* OHRP Guidance on Reviewing and Reporting Unanticipated Problems Involving Risks to Subjects or Others and Adverse Events, January 15, 2007 at http: //www. hhs. gov/ohrp/policy/advevntguid. html#Q 2.
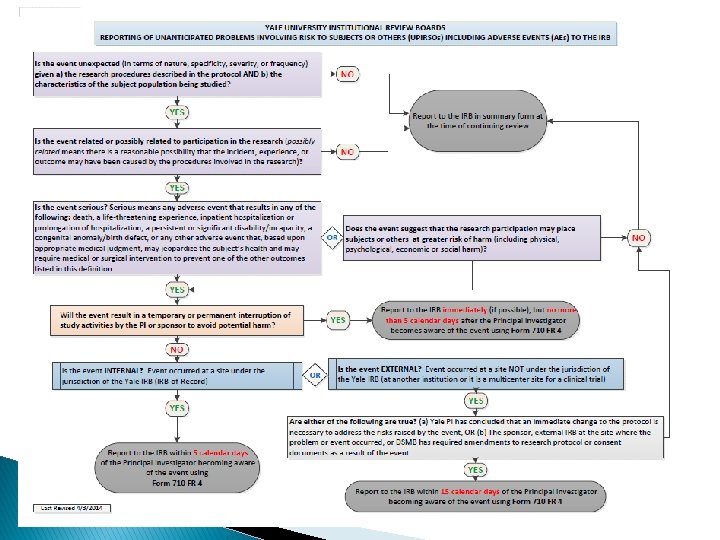

Internal vs. External Internal Event � An event that occurs at a study site under the jurisdiction of a Yale IRB (i. e. , Yale IRB serves as the IRB of record). External Event � An event that occurs at a study site NOT under the jurisdiction of a Yale IRB (e. g. , at another institution in a multicenter clinical trial).

Timeframe for Reporting Internal Events should be reported to the IRB via an RNI in IRES within 5 calendar days of the Principal Investigator becoming aware of the event.
 External Events should be reported to the IRB via an")
Timeframe for Reporting (cont’d) External Events should be reported to the IRB via an RNI in IRES within 15 calendar days of the Yale University Principal Investigator (PI) becoming aware of the event ONLY IF either of the following are true: 1. 2. The Yale PI has concluded that an immediate change to the protocol is necessary to address the risks raised by the event, OR A monitoring entity (e. g. , an external IRB at the site where the problem or event occurred, the sponsor, or the Data Safety Monitoring Board) has required modifications/amendments to the research protocol or consent documents as a result of the event.
 Any events that may require a temporary or permanent interruption")
Timeframe for Reporting (cont’d) Any events that may require a temporary or permanent interruption of study activities by the Principal Investigator or sponsor to avoid potential harm to subjects should be reported to the IRB immediately (if possible), followed by a written report to the IRB via an RNI in IRES no more than 5 calendar days after the Yale Principal Investigator becomes aware of the event.

Case Example #1 � As part of a Phase III, Double-Blind, Placebo- Controlled study, each study participant receives a 30 -minute infusion of the study drug XYZ-123 four times over a period of six weeks. Upon administration of the second infusion, Yale Subject #001 goes into anaphylactic shock requiring immediate hospitalization.

Case Example #1 - Question � Is this Adverse Event reportable to the IRB? ◦ Yes ◦ Not enough information

Review of Case Example #1 � Does the event meet the 3 criteria for reporting promptly? 1. Unexpected; AND 2. Related or Possibly Related; AND 3. Suggests that the research places subjects or others at greater risk of harm (always true if event is a Serious Adverse Event). Answer is “Yes” � Is it an internal or external event? o Internal event given that it involved a Yale subject
 � Is a temporary or permanent interruption of")
Review of Case Example #1 (cont’d) � Is a temporary or permanent interruption of study activities required by the Principal Investigator or sponsor to avoid potential harm to subjects? o If yes, the event should be reported to the IRB immediately (if possible), followed by a written report to the IRB using the UPIRSO Reporting Form within 5 calendar days.

Case Example #2 � A Yale PI receives an IND Safety Report from the Sponsor that indicates that a study subject participating in a phase 3, randomized, double-blind, controlled clinical trial comparing the relative safety and efficacy of a new chemotherapy agent combined with the current standard chemotherapy regime at another site has developed acute pancreatitis requiring hospitalization. � The site PI and the study sponsor have concluded that the acute pancreatitis is possibly related to the study drug. � Neither the consent form, nor the sponsor’s protocol, identify pancreatitis as a risk or potential side effect of the study drug.

Case Example #2 - Question � Is this Adverse Event reportable to the IRB? ◦ Yes ◦ Not enough information

Review of Case Example #2 � Does the event meet the 3 criteria for reporting promptly? 1. Unexpected; AND 2. Related or Possibly Related; AND 3. Suggests that the research places subjects or others at greater risk of harm (always true if event is a Serious Adverse Event). Answer is “Yes” BUT now we need to consider where the event occurred � Is it an internal or external event? o External event – occurred at another study site in a multi -center study
 � External event and therefore, must consider whether")
Review of Case Example #2 (cont’d) � External event and therefore, must consider whether either of the following true? The Yale PI has concluded that an immediate change to the protocol is necessary to address the risks raised by the event; OR o A monitoring entity (e. g. , external IRB for the site where the event occurred, study sponsor or DSMB) has required modification(s)/amendment(s) to the research protocol or consent documents as a result of the event. IF yes, report to IRB within 15 calendar days o

Case Example #3 � A PI conducting behavioral research collects individually identifiable sensitive information about illicit drug use and other illegal behaviors by surveying college students. The data are stored on a laptop and the laptop computer is stolen from the PI’s car on the way home from work.

Case Example #3 - Question � Is this an Unanticipated Problem Involving Risks to Subjects or Others reportable to the IRB? ◦ Yes ◦ Not enough information

Review of Case Example #3 � Does the event meet the 3 criteria for reporting promptly? 1. Unexpected; AND 2. Related or Possibly Related; AND 3. Suggests that the research places subjects or others at greater risk of harm (always true if event is a Serious Adverse Event). Answer is “Yes” � Is it an internal or external event? o Internal event – involved Yale research subjects’ data

Case Example #4 � A subject enrolled in a phase 3, randomized, double-blind, placebo- controlled clinical trial of a new investigational anti-inflammatory agent for management of osteoarthritis develops severe abdominal pain and nausea one month after randomization. Subsequent medical evaluation reveals gastric ulcers. The IRB-approved protocol and informed consent document for the study indicated that there was a 15% chance of developing mild to moderate gastritis and a 2% chance of developing gastric ulcers for subjects assigned to the active investigational agent. � The PI concludes that the subject’s gastric ulcers resulted from the research intervention and withdraws the subject from the study. � A review of data on all subjects enrolled so far reveals that the incidence of gastritis and gastric ulcer are within the expected frequency.

Case Example #4 - Question � Is this Adverse Event reportable to the IRB? ◦ Yes ◦ Not enough information

Review of Case Example #4 � Does the event meet the 3 criteria for reporting promptly? 1. Unexpected; AND 2. Related or Possibly Related; AND 3. Suggests that the research places subjects or others at greater risk of harm (always true if event is a Serious Adverse Event). Answer is “No” This event does not meet the criteria for prompt reporting because the occurrence of gastric ulcers – in terms of nature, severity, and frequency – was expected.

Case Example #5 � As part of an alcohol dependence study, cognitive testing is conducted at each study visit. During one of the study visits, a study participant arrives in an intoxicated state and is unable to complete the cognitive test. The test is rescheduled and the study participant leaves under the escort of a responsible friend after assessment by the researcher that no medical intervention is required.

Case Example #5 - Question � Is this an Unanticipated Problem Involving Risks to Subjects or Others reportable to the IRB? ◦ Yes ◦ Not enough information

Review of Case Example #5 � Does the event meet the 3 criteria for reporting promptly? 1. Unexpected; AND 2. Related or Possibly Related; AND 3. Suggests that the research places subjects or others at greater risk of harm (always true if event is a Serious Adverse Event). Answer is “No” This event does not meet the criteria for prompt reporting because the fact that study participant arrives in an intoxicated state (in an alcohol dependence study) was not unexpected.

Case Example #6 � As required in the study protocol, all study participants on the Phase I study of oral medication ABC-678 for chronic anemia are to be dispensed a two-week supply of study medication. Subject #202 was dispensed the study medication and drove home to his residence. Within an hour of arriving home, the study participant discovers that his four-year old son has ingested some of the study drug ABC-678.

Case Example #6 - Question � Is this an Unanticipated Problem Involving Risks to Subjects or Others reportable to the IRB? ◦ Yes ◦ Not enough information

Review of Case Example #6 � Does the event meet the 3 criteria for reporting promptly? 1. Unexpected; AND 2. Related or Possibly Related; AND 3. Suggests that the research places subjects or others at greater risk of harm (always true if event is a Serious Adverse Event). Answer is “Yes”
- Slides: 32