Amino acid disorders Phenylketonuria PKU Enzyme defect phenylalanine


 • Enzyme defect: phenylalanine hydroxylase (12 th chromosome): more than 400 mutations")
: Variants 1. Classical phenylketonuria (complete or near complete enzyme deficiency): phenylalanine levels")
: Clinical findings • Severe brain damage, progressive motor-mental retardation • Spasticity •")

 and low tyrosine (N: <2")



. tyrosine levels: normal or slightly")

, check")





 •")

 and low")
 polymorphism, thermolabile variant, homozygosity, up to")



,")







 • Hyperammonemia • Hyperlactatemia Diagnosis")


 Biotinidase Biotin (free) piruvate carboxylase propionyl Co. A carboxylase betamethylcrotonyl")


")
 Genetics • Ornitine transcarbabamylase deficiency (most")
 is related to high")
")
")



 Carnitine enzymes Fatty acid (mitochondria) Beta-oxidation (acyl Co.")




 • Life-threatening hypoketotic hypoglycemic coma during catabolic states (prolonged fasting,")

, no")
- Slides: 57
Amino acid disorders
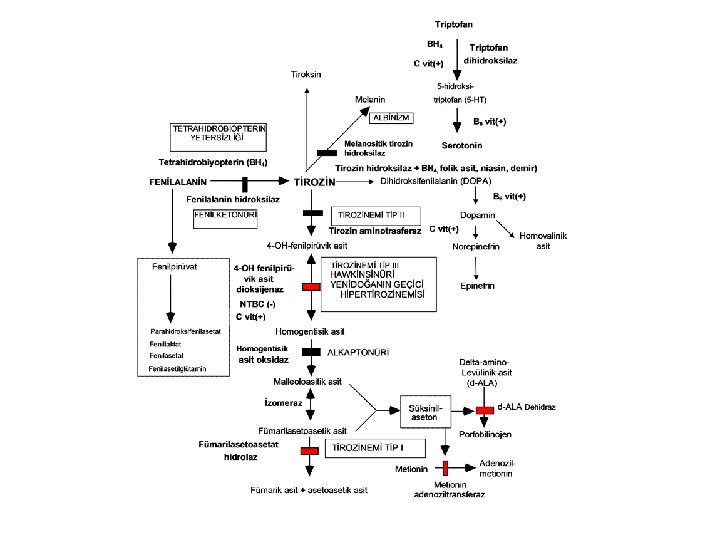
Phenylketonuria (PKU) • Enzyme defect: phenylalanine hydroxylase (12 th chromosome): more than 400 mutations • Incidence: Average 1: 10, 000 (Highest incidence in Turkey, 1: 4, 000)
Phenylketonuria (PKU): Variants 1. Classical phenylketonuria (complete or near complete enzyme deficiency): phenylalanine levels above 20 mg/d. L (<1200 mmol/L) require diet therapy 2. Atypical phenylketonuria (partial enzyme deficiency): (enzyme activity %1 -5) require partial diet therapy 3. Benign phenylketonuria. phenylalanine levels below: 10 mg/d. L (<600 mmol/L) no clinical findings, not requiring diet therapy 3. Malign phenylketonuria: Tetrahydrobiopterin (BH 4=cofactor of phenylalanine hydroxylase): Severe neurologic findings, does not respond diet therapy. Dopamine and setotonin may be helpful.
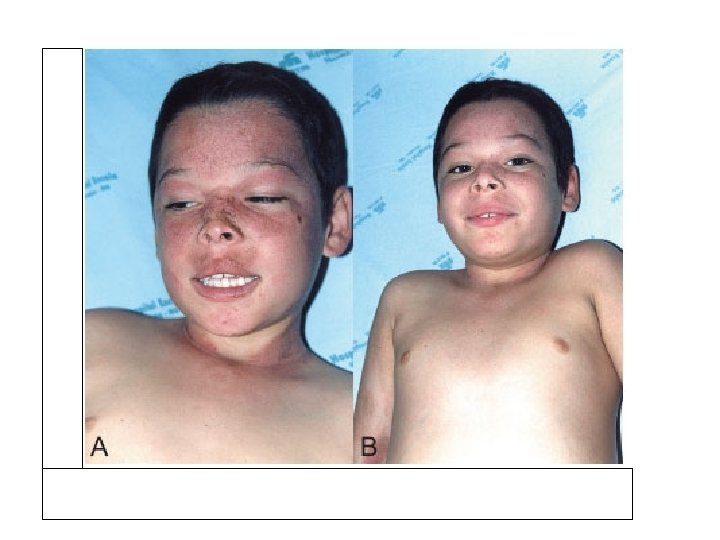
Phenylketonuria (PKU): Clinical findings • Severe brain damage, progressive motor-mental retardation • Spasticity • Self-mutilation • Light colored skin and eye (yellow hair, blue eyes; tyrosine deficiency) • Paralysis • Convulsions • Mouse-like odor in urine and sweat.
Phenylketonuria: Diagnosis • High phenylalanine (N: <2 mg/d. L) and low tyrosine (N: <2 mg/d. L) levels, • Ferric chloride test gives green color in urine (not reliable). • Neonatal screening: Guthrie-card (taken between 3 rd and 7 th days of life)
Phenylketonuria: Therapy • Phenylalanine restricted diet, supplementation of tyrosine, essential amino acids and trace elements. Goals of therapy: • 0 -10 years: phenylalanine values: 0. 7 -4 mg/d. L • 11 -16 years: phenylalanine values: <15 mg/d. L • 16+ years: phenylalanine values: <20 mg/d. L • Pregnant mothers with PKU: phenylalanine values < 7 mg/d. L Prognosis: with immediate and efficient treatment, normal development and intelligence
Maternal PKU= phenylketonuric fetopathy • Normal phenylalanine levels • Microcephaly • Cardiac defects • Motor-mental retardation • No therapy
Tyrosinemia Type I Enzyme defect: Fumarylacetoacetate hydroxylase Clinical findings • Acute infantile form: Severe liver failure, vomiting, bleeds, sepsis, hypoglycemia, renal tubulopathy (Fanconi syndrome) • Chronic form: Hepatomegaly, cirrhosis, growth retardation, rickets, hematoma, tubulopathy, neuropathy, and abdominal pain (due to porphyrines)
Tyrosinemia Type I: Diagnosis • High succinylacetone levels (diagnostic). tyrosine levels: normal or slightly elevated. • Methionine: high • Delta-aminolevulinic acid: high (colic) • Alfa-feto protein: very high (marker of hepatocellular carcinoma)
Tyrosinemia Type I: Therapy • NTBC 1 mg/kg: blocks the accumulation of toxic metabolites (succinylacetone); beware tyrosine elevation and give tyrosine-restricted diet • If this therapy fails consider liver transplantation.
Tyrosinemia Type I: Complications • Renal failure • Hepatocellular carcinoma (monitor alfafeto protein), check periodically liver ultrasongraphy and biopsy. Prognosis: Relatively good under NTBC treatment.
Tyrosinemia Type II • Enzyme defect: Cytosolic tyrosine aminotransferase • Clinical findings: Painful corneal lesions (lacrimation, photophobia, scars), mild mental retardation • Diagnosis: High tyrosine and phenylalanine levels • Therapy: Tyrosine and phenylalaninerestricted diet
Alcaptonuria • Enzyme defect: Homogentisate oxygenase • Clinical findings: black discoloration in urine at acid p. H; mild arthritis in adults • Diagnosis: High homogentisic acid levels in urine • Therapy: Protein-restricted diet? NTBC? • Prognosis: Relatively good without treatment
Methionine metabolism
CLASSICAL HOMOCYSTINURIA • Enzyme defect: Cystationine-ßsynthase • Mechanism: Accumulation of homocysteine (collagen disorder) • Clinical findings: Progressive disease, usually starting with school age. Marfan-like appearance (archnodactyly), progressive myopia (the earliest finding), lens dislocation, epilepsy, mental retardation, osteoporosis, thromboembolism !!!
Marfan syndrom
HOMOCYSTINURIA • Diagnosis: High methionine, high homocysteine (N: 0 -3. 5 µmol/L) and low cysteine levels. Positive nitroprusside test in fresh urine • Therapy: Pyridoxine (Vit. B 6): 50 -1000 mg/day + folic acid 10 mg/day. • If this fails diet + betaine (100 mg/kg) up to 3 X 3 g • Goal: Keep homocysteine <30µmol/L.
MILD HYPERHOMOCYSTEINEMIA Causes • Methylene tetrahydrofolate reductase (MTHFR) polymorphism, thermolabile variant, homozygosity, up to 5% in Europeans, 60% in Asiasns • Heterozygosity for cystationine-ß-synthase • Endogenous and exogenous disorders of folic acid metabolism • Vitamin B 12 deficiency
MILD HYPERHOMOCYSTEINEMIA Clinical findings: • Premature vascular disease in the 3 rd and 4 th decade (infarctions, thrombosis embolies) Maternal hyperhomocysteinemia: congenital defects • Neural tube defects • Cardiac output defects • Renal defects • Pyloric stenosis?
Maple syrup urine disease Enzyme: Branched-chain alfa-ketoacid dehydrogenase complex Incidence: 1: 200, 000, autosomal recessive Clinical findings • Severe form: Progressive encephalopathy, cerebral edema, lethargy, coma after the 3 rd day of life, “çemen” odor in urine and sweat • Mild form: Developmental retardation, recurrent ketoacidotic decompensation
Diagnosis: • “Çemen” odor in urine and sweat, positive DNPH test in urine (non-spesific), • Aminoacid analysis: high valine, leucine, isoleucine and alloisoleucine (diagnostic) levels. Therapy: • Acute: Detoxification (dialysis, exchange transfusion) Augmentation of anabolism : Glucose + insulin • Chronic: Diet (monitor leucine level) ± vitamin B 1 (thiamin): 5 mg/kg/day
Disorders of amino acid transport
Methionine Malabsorption • Methionine malabsorption in renal tubules and intestines. • Clinical findings: White hair, convulsions, , diarrhea, edema , mental retardation, odor (like beer). • Therapy: Diet deficient in methionine.
HARTNUP DISEASE • Defect: Intestinal and renal tubular reabsorption defect of the neutral amino acids (alanine, valine, threonine, leucine, isoleucine, phenylalanine, tyrosine, tryptophan, histidine, glycine; tryptophan deficiency leads nicotinic acid and serotonine deficiency. • Clinical finding: Photodermatitis, cerebellar ataxia; often asymptomatic • Diagnosis: High levels of neutral amino acids in urine low levels of neutral amino acids in plasma. • Therapy: Nicotinamide 40 -300 mg/day, sun protection
LYSINURIC PROTEIN INTOLERANCE • Defect: Intestinal and renal tubular reabsorption defect of the dibasic amino acids (lysine, arginine and ornithine) lead blockage of urea cycle; lysine deficiency • Clinical findings: Intestinal protein intolerance, failure to thrive, osteoporosis, and hyperammonemia with progressive encephalopathy • Diagnosis: Hyperammonemia, low lysine, arginine and ornithine in plasma, high LDH levels. • Therapy: Citrulline substitution, protein restriction
CYSTINURIA • Defect: Renal tubular reabsorption defect of the dibasic amino acids (lysine, arginine, ornithine and cystine) • Clinical findings: Neprolithiasis (cystine crystallizes above 1250 µmol/L at p. H 7. 5) • Diagnosis: Positive nitroprusside test in urine, increased levels of acids lysine, arginine, ornithine and cystine in urine, plasma levels are generally normal. • Therapy: High (>5 L) fluid intake, alkalisation of the urine (urinary infections!). Consider penisillamine (1 -2 g/day), mercaptopropionylglycine or captopril in selected cases.
ORGANIC ACIDEMIAS Pahogenesis • Mitochondrial accumulation of related Co. Ametabolites Clinical findings Acute neonatal form • Lethargy • Feeding problems • Myoclonic jerks • Dehydration * Coma * Hypotonia/hypertonia * Cerebral edema * Unusual odor
ORGANIC ACIDEMIAS: Forms Acute intermittent form • Recurrent episodes of acidotic coma • Ataxia • Focal neurologic signs Chronic progressive form • Failure to thrive, Anorexia • Chronic vomiting • Hypotonia • Developmental retardation
ORGANIC ACIDEMIAS Laboratory findings • Acidosis (increased anion gap) • Hyperammonemia • Hyperlactatemia Diagnosis • Organic acids in urine (GC-MS) • Enzyme and DNA studies
ORGANIC ACIDEMIAS: Therapy Acute • Remove toxins: dialysis, hemofiltration and exchange transfusion • Interrupt catabolic state • Stop protein intake • Give carnitine (100 -300 mg/kg) Long term • Protein restricted diet (special formulas if available) • Carnitine • Vitamins (Vit. B 12, Vit. B 1, Vit. B 2, biotin)
Features of some organic acidemias Izovaleric acidemia Ketoacidosis, dehydration, neutropenia, thromboscytopenia, hyperammonemia, sweety feet odor Propionic acidemia Motor-mental retardation, ketoacidosis, dehydration, neutropenia, thromboscytopenia, hyperammonemia, hipoglycemia Methylmalonic acidemia Motor-mental retardation, ketoacidosis, neutropenia, thromboscytopenia, hyperammonemia, hypoglycemia, response to vit B 12 (+)
Biotinidase deficiency Biotin (complex) Biotinidase Biotin (free) piruvate carboxylase propionyl Co. A carboxylase betamethylcrotonyl Co. A carboxylase asetyl Co. A carboxylase
Biotinidase deficiency Incidense World. 1: 60, 000 Turkey: 1: 10, 000 Clinical and laboratory findings • Severe metabolic acidosis • Alopecia • Seborrheic skin eruptions • Refractory convulsions Therapy 5 -10 mg/day biotin (life long).
Urea cycle defects
Carbaglu (+)
Urea cycle defects Incidence: 1: 10, 000 (cumulative) Genetics • Ornitine transcarbabamylase deficiency (most common urea cycle defect, X-linked) • Argininosuccinate synthase deficiency (citrullinemia, (the second most common urea cycle defect, OR) • Carbamylphosphate synthase I deficiency (OR) • Argininosuccinate lyase deficiency (argininosuccinic aciduria, OR) • Arginase deficiency (argininemia, OR)
Urea cycle defects: Clinical findings Main symptom (acute/or chronic encephalopathy) is related to high protein intake, increased catabolism, infections or stress Neonates: * Poor feeding * Lethargy * Loss of reflexes * Seizures * Temperature lability * Hyperventilation (respiratory alkalosis) * Intracranial hemorrhages * Progressive encephalopathy Infants and children * Failure to thrive * Feeding problems * Nausea, vomiting * Episodic encephalopathy * Ataxia * Convulsions Adolescents and adults * Chronic neurologic symptoms * Chronic psychiatric symptoms * Episodic encephalopathy * Behavioral problems
Urea cycle defects Laboratory findings • Hyperammonemia (generally >400 µmol/L in urea cycle defects) • Amino acids in serum • Organic acids in urine Differential diagnosis • Organic acidurias: • Liver diseases: neonatal hepatitis, galactosemia, tyrosinemia, respiratory chain defects • Transient hyperammonemia of newborn due to patent ductus venosus.
CPS= Karbamoil fosfat sentaz OTC= Ornitin transkarbomoilaz ASA=Arjininosüksinik asit AS=Arjininosüksinat sentaz AL=Arjininosüksinat liaz(sitrüllinemi)
Urea cycle defects: Acute therapy • Stop protein intake • Interrupt catabolic state by high calorie infusion (carbohydrate + lipid) • Remove ammonia when >400 µmol/L by hemodiafiltration, hemofiltration, or hemodialysis, (periton dialysis is not effective) • Give arginine 350 mg/kg in order to support urea cycle. • Give sodium benzoate: 350 mg/kg/day • Give sodium phenylbutyrate 250 mg/kg/day • Aim for an ammonia concentration < 200µmol/L
Urea cycle defects Chronic therapy • Restriction of protein intake (1. 0 -1. 5 g/kg/day) +arginine + • sodium benzoate + sodium –phenylbutyrate Prognosis • Poor if there is prolonged coma (>3 days), and symptoms and signs of increased intracranial pressure
Defects of Fatty acid oxidation
Fatty acid oxidation Fatty acid (plasma) Carnitine enzymes Fatty acid (mitochondria) Beta-oxidation (acyl Co. A dehydrogenases) Krebs cycle Asetil Co. A 131 ATP 3 -ketothiolase (tioforase) Keton bodies HMG Co. A- liase HMG Co. Asynthase
Fatty acid oxidation
• Disorders of fatty acid oxidation • During prolonged fasting mitochonrial oxidation of fatty acids provides up to 80% of the total energy requirement.
Fatty acid oxidation: Etiology Carnitine transporter deficiency Defects of carnitine cycle • Carnitine palmitoyltransferase I (CPTI) deficiency • Carnitine translocase deficiency • Carnitine palmitoyltransferase II (CPTII) deficiency ß-oxidation defects • Very long-chain acyl-Co. A dehydrogenase (VLCAD) deficiency • Medium-chain acyl-Co. A dehydrogenase (MCAD) deficiency • Short-chain acyl-Co. A dehydrogenase (SCAD) deficiency • Long-chain hydroxyacyl-Co. A dehydrogenase (LCHAD) deficiency • Medium-chain hydroxyacyl-Co. A dehydrogenase (LCHAD) deficiency • Short-chain hydroxyacyl-Co. A dehydrogenase (LCHAD) deficiency
Fatty acid oxidation: Pathogenesis • Insufficient energy production during fasting • Deficiency of mitochondrial free Co. A due to accumulation of toxic intermediary products
Clinical findings (Reyelike syndrome) • Life-threatening hypoketotic hypoglycemic coma during catabolic states (prolonged fasting, infections, operations) • Liver failure • Skeletal myopathy, cardiomyopathy
Fatty acid oxidation: Laboratory findings • Ketones: low, ammonia: high, glucose: low to normal, liver enzymes: high • Total carnitine: low (high in CPTI deficiency) • Acyl carnitine/total carnitine: Low • Dicarboxilic acids in urine (GS-MS) • Acylcarnitine profile (Diagnostic) • Enzyme studies (Fibroblasts, lymphocytes)
Fatty acid oxidation: therapy Acute therapy • High dose glucose (7 -10 mg/kg/min), no lipids (!) • Carnitine (100 mg/kg): not in carnitine cylce defects, and in LCHAD deficiency Chronic therapy • Avoid prolonged fasting, careful monitoring during catabolic states