Advanced BLAST Searching Courtesy of Jonathan Pevsner Johns

Advanced BLAST Searching Courtesy of Jonathan Pevsner Johns Hopkins U.

Outline of today’s lecture Organism-specific BLAST sites Specialized BLAST-related algorithms BLAST-like tools for genomic DNA searches PSI-BLAST PHI-BLAST Find-a-gene

Specialized BLAST servers Organism-specific BLAST sites
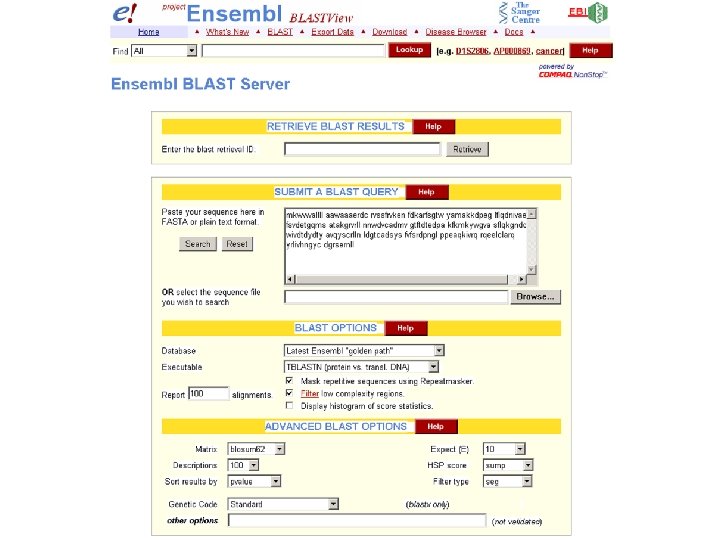

Ensembl BLAST output includes an ideogram

TIGR BLAST
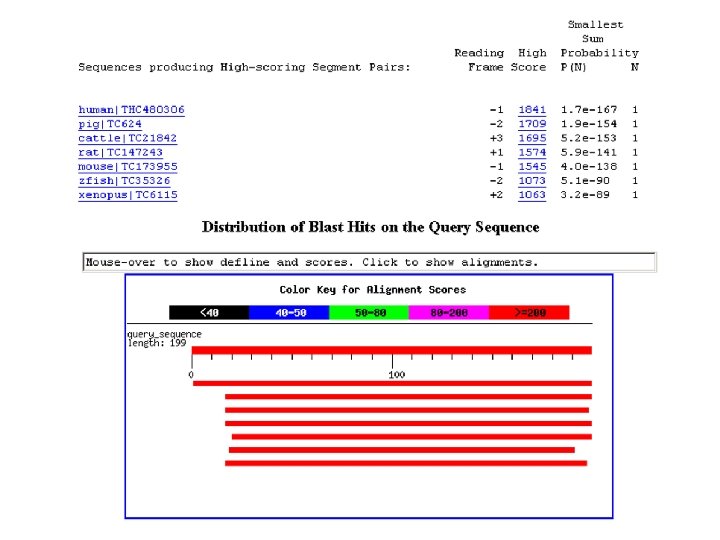
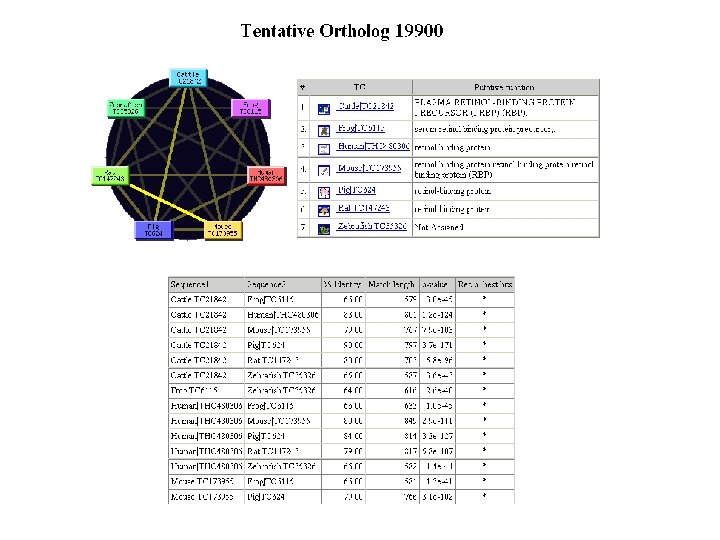

BLAST output of Pro. Dom server: graphical view of domains
")
Specialized BLAST servers Species-specific BLAST sites Molecule-specific BLAST sites Specialized algorithms (WU-BLAST 2. 0)
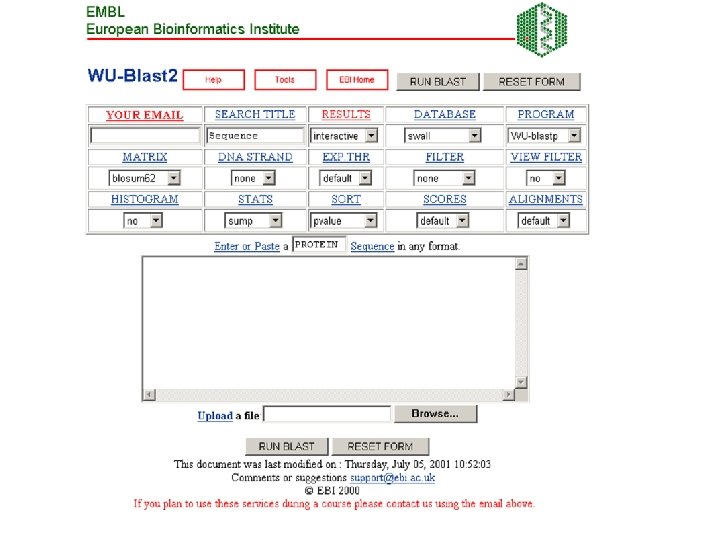
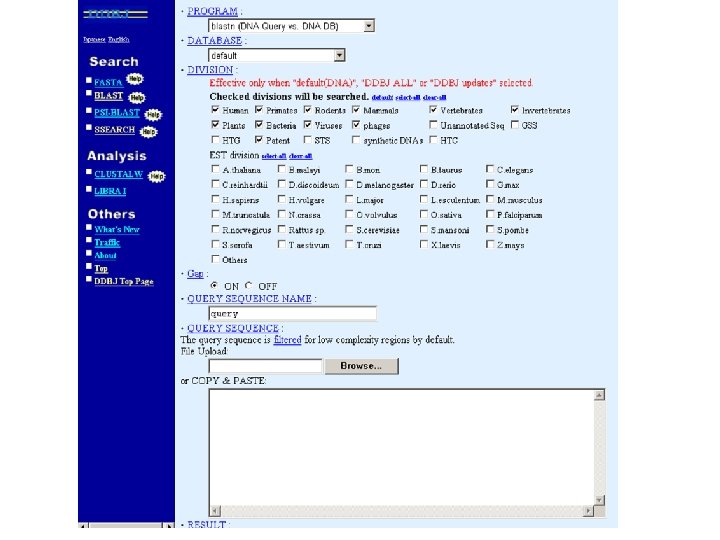

FASTA server at the University of Virginia
")
Conserved Domain Database (CDD)
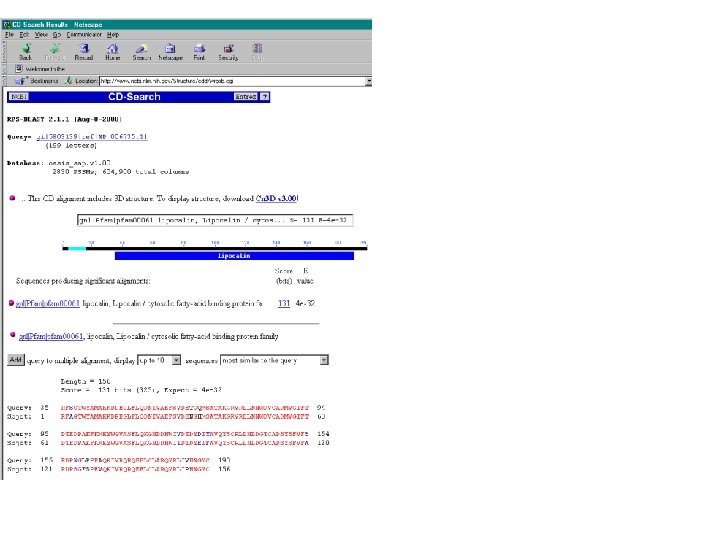
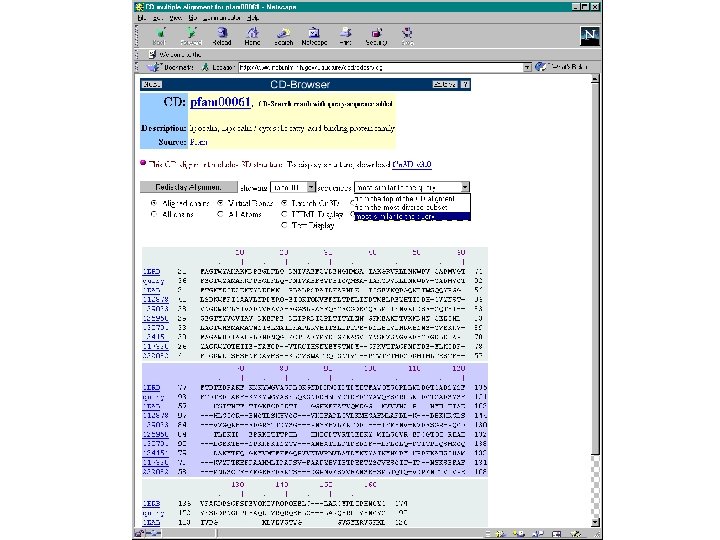
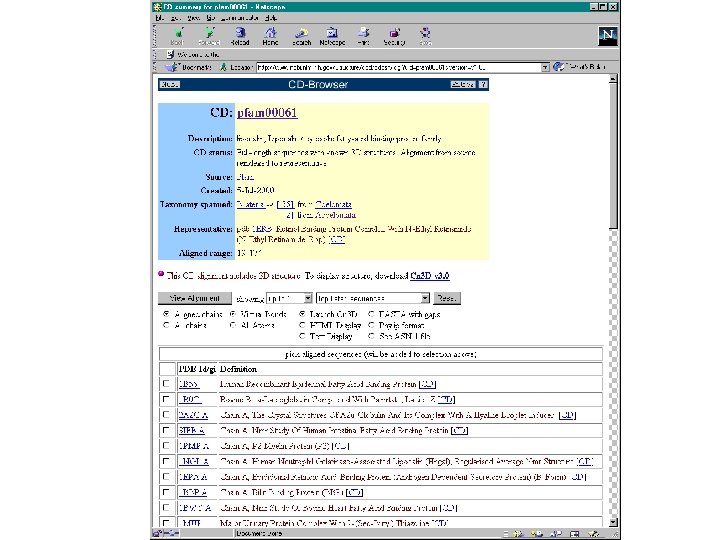

BLAST-related tools for genomic DNA The analysis of genomic DNA presents special challenges: • There are exons (protein-coding sequence) and introns (intervening sequences). • There may be sequencing errors or polymorphisms • The comparison may be between related species (e. g. human and mouse)

BLAST-related tools for genomic DNA Recently developed tools include: • Mega. BLAST at NCBI. • BLAT (BLAST-like alignment tool). BLAT parses an entire genomic DNA database into words (11 mers), then searches them against a query. Thus it is a mirror image of the BLAST strategy. See http: //genome. ucsc. edu • SSAHA at Ensembl uses a similar strategy as BLAT. See http: //www. ensembl. org

To access BLAT, visit http: //genome. ucsc. edu

Paste DNA or protein sequence here in the FASTA format

BLAT output includes browser and other formats

Position specific iterated BLAST: PSI-BLAST The purpose of PSI-BLAST is to look deeper into the database for matches to your query protein sequence by employing a scoring matrix that is customized to your query.
![PSI-BLAST is performed in five steps [1] Select a query and search it against](http://slidetodoc.com/presentation_image/5d5454dc7233868b76e20dc5878fcd86/image-24.jpg "PSI-BLAST is performed in five steps [1] Select a query and search it against")
PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database
![PSI-BLAST is performed in five steps [1] Select a query and search it against](http://slidetodoc.com/presentation_image/5d5454dc7233868b76e20dc5878fcd86/image-25.jpg "PSI-BLAST is performed in five steps [1] Select a query and search it against")
PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database [2] PSI-BLAST constructs a multiple sequence alignment then creates a “profile” or specialized position-specific scoring matrix (PSSM)

R, I, K C D, E, T K, R, T N, L, Y, G

1 M 2 K 3 W 4 V 5 W 6 A 7 L 8 L 9 L 10 L 11 A 12 A 13 W 14 A 15 A 16 A. . . 37 S 38 G 39 T 40 W 41 Y 42 A A -1 -1 -3 0 -3 5 -2 -1 -1 -2 5 5 -2 3 2 4 R -2 1 -3 -3 -3 -2 -2 -2 -3 -2 -1 -2 N -2 0 -4 -3 -4 -2 -4 -3 -4 -4 -2 -2 -4 -1 0 -1 D -3 1 -5 -4 -5 -2 -4 -4 -2 -2 -4 -2 -1 -2 C -2 -4 -3 -1 -1 -2 -1 Q -1 2 -2 -3 -2 -1 -2 -3 -2 -2 -1 -1 -2 -1 E -2 4 -3 -3 -3 -1 -1 -3 -2 0 -1 G -3 -2 -3 -4 -3 0 -4 -4 0 0 -4 4 2 3 H -2 0 -3 -4 -3 -2 -3 -3 -2 -2 -3 -2 -1 -2 I 1 -3 -3 -2 2 2 -2 -2 1 -2 -3 -2 L 2 -3 -2 1 -2 -2 4 4 -2 -2 4 -2 -3 -2 K -2 3 -3 -3 -3 -1 -1 -3 -1 0 -1 M 6 -2 -2 1 -2 -1 2 2 -1 -1 2 -2 -2 -1 F 0 -4 1 -1 1 -3 0 0 -3 -3 1 -3 -3 -3 P -3 -1 -4 -3 -4 -1 -3 -3 -1 -1 -1 S -2 0 -3 -2 -3 1 -3 -2 -3 -3 1 1 -3 1 T -1 -1 -3 0 -1 -1 0 0 -2 -1 0 0 W -2 -3 12 -3 -2 -2 -3 -3 7 -3 -3 -3 Y -1 -2 2 -1 2 -2 -1 0 -1 -1 -2 -2 0 -3 -2 -2 V 1 -3 -3 4 -3 0 1 3 2 1 0 0 0 -1 -2 -1 2 0 0 -3 -2 4 -1 -3 -2 -2 0 -1 0 -4 -2 -2 -1 -5 -3 -2 -1 -3 -3 -1 0 -2 -1 -2 -2 -1 0 0 -1 -2 -3 -2 6 -2 -4 -4 -1 -2 -2 -1 -1 -3 -3 -2 -2 -3 2 -2 -1 -1 0 -2 -2 -2 0 -2 -1 -3 -2 -1 -2 -3 -1 -2 -1 -1 -3 -4 -2 1 3 -3 -1 4 1 -3 -2 0 -2 -3 -1 1 5 -3 -4 -3 -3 12 -3 -2 -2 2 -1 1 0 -3 -2 2 7 -2 -2 -4 0 -3 -1 0

1 M 2 K 3 W 4 V 5 W 6 A 7 L 8 L 9 L 10 L 11 A 12 A 13 W 14 A 15 A 16 A. . . 37 S 38 G 39 T 40 W 41 Y 42 A A -1 -1 -3 0 -3 5 -2 -1 -1 -2 5 5 -2 3 2 4 R -2 1 -3 -3 -3 -2 -2 -2 -3 -2 -1 -2 N -2 0 -4 -3 -4 -2 -4 -3 -4 -4 -2 -2 -4 -1 0 -1 D -3 1 -5 -4 -5 -2 -4 -4 -2 -2 -4 -2 -1 -2 C -2 -4 -3 -1 -1 -2 -1 Q -1 2 -2 -3 -2 -1 -2 -3 -2 -2 -1 -1 -2 -1 E -2 4 -3 -3 -3 -1 -1 -3 -2 0 -1 G -3 -2 -3 -4 -3 0 -4 -4 0 0 -4 4 2 3 H -2 0 -3 -4 -3 -2 -3 -3 -2 -2 -3 -2 -1 -2 I 1 -3 -3 -2 2 2 -2 -2 1 -2 -3 -2 L 2 -3 -2 1 -2 -2 4 4 -2 -2 4 -2 -3 -2 K -2 3 -3 -3 -3 -1 -1 -3 -1 0 -1 M 6 -2 -2 1 -2 -1 2 2 -1 -1 2 -2 -2 -1 F 0 -4 1 -1 1 -3 0 0 -3 -3 1 -3 -3 -3 P -3 -1 -4 -3 -4 -1 -3 -3 -1 -1 -1 S -2 0 -3 -2 -3 1 -3 -2 -3 -3 1 1 -3 1 T -1 -1 -3 0 -1 -1 0 0 -2 -1 0 0 W -2 -3 12 -3 -2 -2 -3 -3 7 -3 -3 -3 Y -1 -2 2 -1 2 -2 -1 0 -1 -1 -2 -2 0 -3 -2 -2 V 1 -3 -3 4 -3 0 1 3 2 1 0 0 0 -1 -2 -1 2 0 0 -3 -2 4 -1 -3 -2 -2 0 -1 0 -4 -2 -2 -1 -5 -3 -2 -1 -3 -3 -1 0 -2 -1 -2 -2 -1 0 0 -1 -2 -3 -2 6 -2 -4 -4 -1 -2 -2 -1 -1 -3 -3 -2 -2 -3 2 -2 -1 -1 0 -2 -2 -2 0 -2 -1 -3 -2 -1 -2 -3 -1 -2 -1 -1 -3 -4 -2 1 3 -3 -1 4 1 -3 -2 0 -2 -3 -1 1 5 -3 -4 -3 -3 12 -3 -2 -2 2 -1 1 0 -3 -2 2 7 -2 -2 -4 0 -3 -1 0
![PSI-BLAST is performed in five steps [1] Select a query and search it against](http://slidetodoc.com/presentation_image/5d5454dc7233868b76e20dc5878fcd86/image-29.jpg "PSI-BLAST is performed in five steps [1] Select a query and search it against")
PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database [2] PSI-BLAST constructs a multiple sequence alignment then creates a “profile” or specialized position-specific scoring matrix (PSSM) [3] The PSSM is used as a query against the database [4] PSI-BLAST estimates statistical significance (E values)

![PSI-BLAST is performed in five steps [1] Select a query and search it against](http://slidetodoc.com/presentation_image/5d5454dc7233868b76e20dc5878fcd86/image-31.jpg "PSI-BLAST is performed in five steps [1] Select a query and search it against")
PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database [2] PSI-BLAST constructs a multiple sequence alignment then creates a “profile” or specialized position-specific scoring matrix (PSSM) [3] The PSSM is used as a query against the database [4] PSI-BLAST estimates statistical significance (E values) [5] Repeat steps [3] and [4] iteratively, typically 5 times. At each new search, a new profile is used as the query.

Results of a PSI-BLAST search Iteration 1 2 3 4 5 6 7 8 # hits 104 173 236 301 344 342 378 382 # hits > threshold 49 96 178 240 283 298 310 320
,")
PSI-BLAST alignment of RBP and b-lactoglobulin: iteration 1 Score = 46. 2 bits (108), Expect = 2 e-04 Identities = 40/150 (26%), Positives = 70/150 (46%), Gaps = 37/150 (24%) Query: 27 Sbjct: 33 Query: 87 Sbjct: 83 VKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVC 86 V+ENFD ++ G WY + +K P + I A +S+ E G + K ++ VQENFDVKKYLGRWYEI-EKIPASFEKGNCIQANYSLMENGNIEVLNK-----ELS 82 ADMVGTF-----TDTEDPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCR 137 D GT ++ +PAK +++++ + +WI+ TDY+ YA+ YSC PD--GTMNQVKGEAKQSNVSEPAKLEVQFFPLMP-----PAPYWILATDYENYALVYSCT 135 Query: 138 ----LLNLDGTCADSYSFVFSRDPNGLPPE 163 L ++D + ++ R+P LPPE Sbjct: 136 TFFWLFHVD------FFWILGRNPY-LPPE 158
, Expect")
PSI-BLAST alignment of RBP and b-lactoglobulin: iteration 2 Score = 140 bits (353), Expect = 1 e-32 Identities = 45/176 (25%), Positives = 78/176 (43%), Gaps = 33/176 (18%) Query: 4 Sbjct: 2 Query: 56 Sbjct: 61 VWALLLLAAWAAAERDCRVSSF----RVKENFDKARFSGTWYAMAKKDPEGLFLQD 55 V L+ LA A + +F V+ENFD ++ G WY + +K P + VTMLMFLATLAGLFTTAKGQNFHLGKCPSPPVQENFDVKKYLGRWYEI-EKIPASFEKGN 60 NIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMV---GTFTDTEDPAKFKMKYWGVASF 112 I A +S+ E G + K + D + V ++ +PAK +++++ + CIQANYSLMENGNIEVLNKEL-----SPDGTMNQVKGEAKQSNVSEPAKLEVQFFPL--- 112 Query: 113 LQKGNDDHWIVDTDYDTYAVQYSCR----LLNLDGTCADSYSFVFSRDPNGLPPEA 164 +WI+ TDY+ YA+ YSC L ++D + ++ R+P LPPE Sbjct: 113 --MPPAPYWILATDYENYALVYSCTTFFWLFHVD------FFWILGRNPY-LPPET 159
, Expect")
PSI-BLAST alignment of RBP and b-lactoglobulin: iteration 3 Score = 159 bits (404), Expect = 1 e-38 Identities = 41/170 (24%), Positives = 69/170 (40%), Gaps = 19/170 (11%) Query: 3 Sbjct: 1 Query: 55 Sbjct: 60 WVWALLLLAAWAAAERD----CRVSSFRVKENFDKARFSGTWYAMAKKDPEGLFLQ 54 V L+ LA A + S V+ENFD ++ G WY + K MVTMLMFLATLAGLFTTAKGQNFHLGKCPSPPVQENFDVKKYLGRWYEIEKIPASFE-KG 59 DNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTEDPAKFKMKYWGVASFLQ 114 + I A +S+ E G + K V + ++ +PAK +++++ + NCIQANYSLMENGNIEVLNKELSPDGTMNQVKGE--AKQSNVSEPAKLEVQFFPL----- 112 Query: 115 KGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADSYSFVFSRDPNGLPPEA 164 +WI+ TDY+ YA+ YSC + ++ R+P LPPE Sbjct: 113 MPPAPYWILATDYENYALVYSCTTFFWL--FHVDFFWILGRNPY-LPPET 159
, Expect = 2 e-04 Identities = 40/150")
1 Score = 46. 2 bits (108), Expect = 2 e-04 Identities = 40/150 (26%), Positives = 70/150 (46%), Gaps = 37/150 (24%) Query: 27 Sbjct: 33 Query: 87 Sbjct: 83 VKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVC 86 V+ENFD ++ G WY + +K P + I A +S+ E G + K ++ VQENFDVKKYLGRWYEI-EKIPASFEKGNCIQANYSLMENGNIEVLNK-----ELS 82 ADMVGTF-----TDTEDPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCR 137 D GT ++ +PAK +++++ + +WI+ TDY+ YA+ YSC PD--GTMNQVKGEAKQSNVSEPAKLEVQFFPLMP-----PAPYWILATDYENYALVYSCT 135 Query: 138 ----LLNLDGTCADSYSFVFSRDPNGLPPE 163 L ++D + ++ R+P LPPE Sbjct: 136 TFFWLFHVD------FFWILGRNPY-LPPE 158 3 Score = 159 bits (404), Expect = 1 e-38 Identities = 41/170 (24%), Positives = 69/170 (40%), Gaps = 19/170 (11%) Query: 3 Sbjct: 1 Query: 55 Sbjct: 60 WVWALLLLAAWAAAERD----CRVSSFRVKENFDKARFSGTWYAMAKKDPEGLFLQ 54 V L+ LA A + S V+ENFD ++ G WY + K MVTMLMFLATLAGLFTTAKGQNFHLGKCPSPPVQENFDVKKYLGRWYEIEKIPASFE-KG 59 DNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTEDPAKFKMKYWGVASFLQ 114 + I A +S+ E G + K V + ++ +PAK +++++ + NCIQANYSLMENGNIEVLNKELSPDGTMNQVKGE--AKQSNVSEPAKLEVQFFPL----- 112 Query: 115 KGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADSYSFVFSRDPNGLPPEA 164 +WI+ TDY+ YA+ YSC + ++ R+P LPPE Sbjct: 113 MPPAPYWILATDYENYALVYSCTTFFWL--FHVDFFWILGRNPY-LPPET 159
 retinol-binding protein apolipoprotein D odorant-binding")
The universe of lipocalins (each dot is a protein) retinol-binding protein apolipoprotein D odorant-binding protein
 picture retinol-binding protein your")
Scoring matrices let you focus on the big (or small) picture retinol-binding protein your RBP query
 picture PAM 250 PAM")
Scoring matrices let you focus on the big (or small) picture PAM 250 PAM 30 retinol-binding protein Blosum 80 Blosum 45

PSI-BLAST generates scoring matrices more powerful than PAM or BLOSUM retinol-binding protein

PSI-BLAST: performance assessment Evaluate PSI-BLAST results using a database in which protein structures have been solved and all proteins in a group share < 40% amino acid identity.

PSI-BLAST: the problem of corruption PSI-BLAST is useful to detect weak but biologically meaningful relationships between proteins. The main source of false positives is the spurious amplification of sequences not related to the query. For instance, a query with a coiled-coil motif may detect thousands of other proteins with this motif that are not homologous. Once even a single spurious protein is included in a PSI-BLAST search above threshold, it will not go away.

PSI-BLAST: the problem of corruption Corruption is defined as the presence of at least one false positive alignment with an E value < 10 -4 after five iterations. Three approaches to stopping corruption: [1] Apply filtering of biased composition regions [2] Adjust E value from 0. 001 (default) to a lower value such as E = 0. 0001. [3] Visually inspect the output from each iteration. Remove suspicious hits by unchecking the box.

: create an artificial protein consisting of RBP 4")
See problem 5 -1 (page 153): create an artificial protein consisting of RBP 4 and protein kinase C. How does PSI-BLAST perform?


PHI-BLAST: Pattern hit initiated BLAST Launches from the same page as PSI-BLAST Combines matching of regular expressions with local alignments surrounding the match.

PHI-BLAST: Pattern hit intiated BLAST Launches from the same page as PSI-BLAST Combines matching of regular expressions with local alignments surrounding the match. Given a protein sequence S and a regular expression pattern P occurring in S, PHI-BLAST helps answer the question: What other protein sequences both contain an occurrence of P and are homologous to S in the vicinity of the pattern occurrences? PHI-BLAST may be preferable to just searching for pattern occurrences because it filters out those cases where the pattern occurrence is probably random and not indicative of homology.
 ecblc vc hsrbp 1 50 MRLLPLVAAA")
Align three lipocalins (RBP and two bacterial lipocalins) ecblc vc hsrbp 1 50 MRLLPLVAAA TAAFLVVACS SPTPPRGVTV VNNFDAKRYL GTWYEIARFD MRAIFLILCS V. . . LLNGCL G. . MPESVKP VSDFELNNYL GKWYEVARLD ~~~MKWVWAL LLLAAWAAAE RDCRVSSFRV KENFDKARFS GTWYAMAKKD

Pick a small, conserved region and see which amino acid residues are used ecblc vc hsrbp 1 50 MRLLPLVAAA TAAFLVVACS SPTPPRGVTV VNNFDAKRYL GTWYEIARFD MRAIFLILCS V. . . LLNGCL G. . MPESVKP VSDFELNNYL GKWYEVARLD ~~~MKWVWAL LLLAAWAAAE RDCRVSSFRV KENFDKARFS GTWYAMAKKD GTWYEI K AV M

Create a pattern using the appropriate syntax ecblc vc hsrbp 1 50 MRLLPLVAAA TAAFLVVACS SPTPPRGVTV VNNFDAKRYL GTWYEIARFD MRAIFLILCS V. . . LLNGCL G. . MPESVKP VSDFELNNYL GKWYEVARLD ~~~MKWVWAL LLLAAWAAAE RDCRVSSFRV KENFDKARFS GTWYAMAKKD GTWYEI K AV M GXW[YF][EA][IVLM]
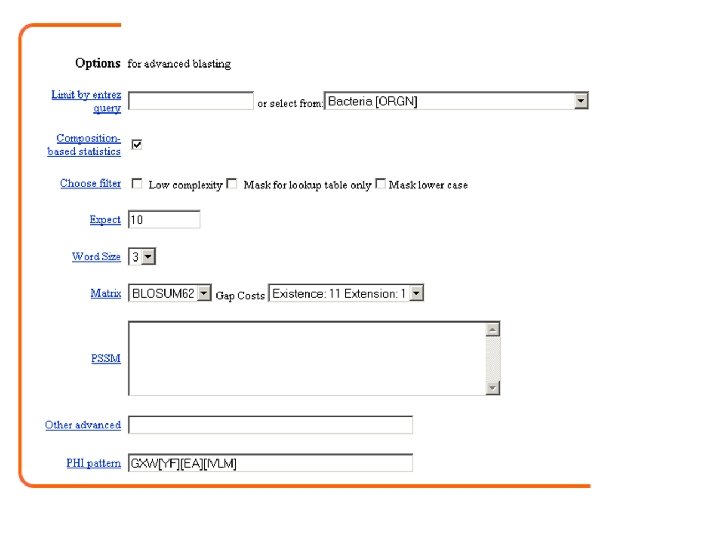
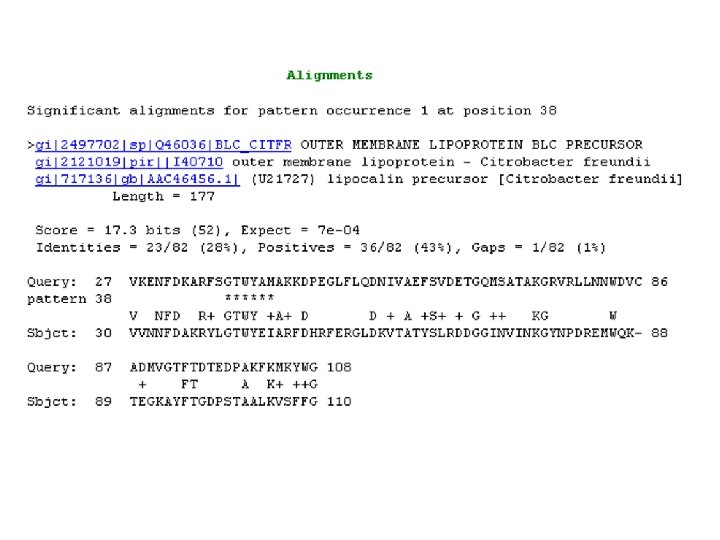

Syntax rules for PHI-BLAST The syntax for patterns in PHI-BLAST follows the conventions of PROSITE (protein lecture, Chapter 8). When using the stand-alone program, it is permissible to have multiple patterns. When using the Web-page only one pattern is allowed per query. [ ] means any one of the characters enclosed in the brackets e. g. , [LFYT] means one occurrence of L or F or Y or T - means nothing (spacer character) x(5) means 5 positions in which any residue is allowed x(2, 4) means 2 to 4 positions where any residue is allowed

BLAST for gene discovery You can use BLAST to find a “novel” gene

Start with the sequence of a known protein tblastn Search a DNA database (e. g. HTGS, db. EST, or genomic sequence from a specific organism) inspect Search your DNA or protein against a protein database (nr) to confirm you have identified a novel gene blastx or blastp nr Find matches… [1] to DNA encoding known proteins [2] to DNA encoding related (novel!) proteins [3] to false positives

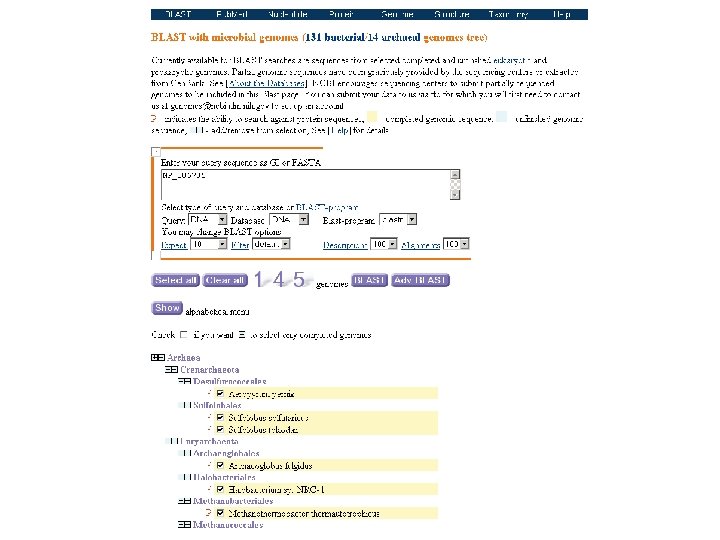


this is a good candidate for a novel gene/protein

A blastp nr search confirms that the Salmonella query is closely related to other lipocalins
- Slides: 61