3 CORSO REGIONALE DI FARMACOVIGILANZA LA FARMACOVIGILANZA ATTIVA

















 Dal 1° ottobre 2008 la")
 La pubblicazione degli avvisi non")
 Misura intrapresa dal fabbricante")



 • Assicura il rispetto dell’iter")


 Mandatario (nome,")










- Slides: 38
3° CORSO REGIONALE DI FARMACOVIGILANZA LA FARMACOVIGILANZA ATTIVA NELLA REGIONE EMILIA-ROMAGNA Bologna 19 novembre 2009 Il ruolo del Referente Aziendale della vigilanza: il punto di vista del Farmacista Ospedaliero Dott. ssa Silvia Galassi Responsabile Vigilanza Dispositivi Medici AUSL di Ravenna
OBIETTIVO DELLA VIGILANZA • migliorare la protezione della salute e della sicurezza dei pazienti e degli utilizzatori, riducendo la probabilità che lo stesso tipo di INCIDENTE si ripeta in posti diversi in tempi successivi, attraverso la condivisione delle informazioni fra Autorità Competente, Fabbricanti e Utilizzatori rendendo quanto più tempestiva l’applicazione delle azioni correttive
NORMATIVA DI RIFERIMENTO – – Art. 11 del D. Lgs 507/92 relativo ai AIMD Art. 9 del D. Lgs 46/97 relativo ai DM Art. 11 del D. Lgs 332/00 relativo ai IVD Circolare del Md. S del 27. 07. 2004 “vigilanza sugli incidenti “ – Decreto 15. 11. 2005 “Approvazione dei modelli di schede di segnalazione” – Linea Guida sul sistema di vigilanza dei DM MEDDEV 2. 12 -1 rev. 5, aprile 2007
ATTORI DELLA VIGILANZA • Il fabbricante deve garantire sicurezza, affidabilità, durata, istruzioni corrette, destinazione d’uso, rintracciabilità. • Il farmacista deve garantire controllo della qualità del prodotto per tutta la fornitura, formazione/informazione, monitoraggio sul corretto utilizzo, rintracciabilità. • L’operatore sanitario deve garantire conservazione corretta, utilizzo secondo scheda tecnica.
ARTICOLO 4 D. Lgs. 46/97 Requisiti essenziali dei DM • “ I dispositivi devono soddisfare i requisiti essenziali prescritti nell’allegato 1 in considerazione della loro destinazione” • Allegato 1 “ 1. I dispositivi devono essere progettati e fabbricati in modo che la loro utilizzazione non comprometta lo stato clinico e la sicurezza dei pazienti , né la sicurezza e la salute degli utilizzatori ed eventualmente di terzi quando siano utilizzati alle condizioni e per fini previsti …
. . . ALLEGATO 1 • 13. Informazioni fornite dal fabbricante • 13. 1 “ Ogni dispositivo deve essere corredato dalle necessarie informazioni per garantire un’utilizzazione sicura e per consentire di identificare il fabbricante…” Le informazioni sono costituite dalle indicazioni riportate sull’etichetta e dalle indicazioni contenute nelle istruzioni per l’uso. Le informazioni necessarie per garantire un’utilizzazione sicura del dispositivo devono figurare sul dispositivo stesso e/o sull’imballaggio unitario o eventualmente sull’imballaggio commerciale.
ARTICOLO 5 D. LGS 46/97 Campo di applicazione • 4. Le indicazioni fornite dal fabbricante all’utilizzatore e al paziente sono espresse in lingua italiana al momento della consegna all’utilizzatore finale, per uso professionale o per qualsiasi altra utilizzazione.
……ALLEGATO 1 • 13. 3 L’etichettatura deve contenere le informazioni seguenti : • nome o ragione sociale e indirizzo del fabbricante …; • le indicazioni necessarie per consentire all’utilizzatore di identificare il dispositivo e il contenuto della confezione; • se del caso, la parola STERILE ; • se del caso, il numero di codice del lotto preceduto dalla parola LOTTO o il numero di serie; • se del caso, l’indicazione della data entro cui il dispositivo dovrebbe essere utilizzato in condizioni di sicurezza , espressa in anno/mese ; • se del caso, l’indicazione che il dispositivo è monouso ; • per i dispositivi su misura , l’indicazione dispositivo su misura ; • per i dispositivi destinati ad indagini cliniche, l’indicazione destinato esclusivamente ad indagini cliniche ; • le condizioni specifiche di conservazione e/o di manipolazione ; • eventuali istruzioni specifiche di utilizzazione; • avvertenze e/o precauzioni da prendere; • metodo di sterilizzazione se del caso.
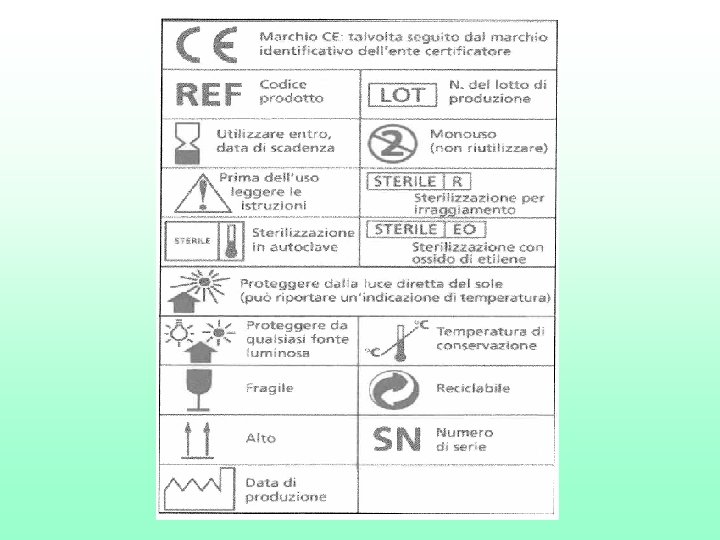
SIMBOLI GRAFICI • La norma tecnica EN 980 propone l’utilizzo di una serie di simboli grafici in sostituzione di intere frasi o concetti volti a spiegare alcune funzioni, a dare delle avvertenze riguardanti i DM. L’obiettivo della norma e’ quello di aiutare a ridurre le molteplici traduzioni di parole nelle lingue nazionali per semplificare l’etichettatura, per rendere disponibili informazioni importanti agli utilizzatori.
ARTICOLO 8 D. LGS 46/97 Classificazione • I dispositivi sono suddivisi in 4 classi : classi I, II a, II b, III. La classificazione segue le regole di cui all’Allegato IX • La suddivisione in 4 classi differenti è in funzione di ben precisi criteri quali la vulnerabilità nell’organismo, la destinazione, il tempo di contatto con l’organismo, i rischi potenziali durante l’impiego, l’elaborazione tecnologica e la fabbricazione. • Alla classe I appartengono i dispositivi non invasivi , cioè quelli che non entrano in contatto né interagiscono con il corpo ; alla classe II a appartengono i dispositivi invasivi a breve termine in orifizi naturali del corpo e quelli invasivi chirurgici per uso temporaneo; alla classe II b appartengono quelli invasivi chirurgici a breve termine, mentre la classe III include quelli destinati ad entrare in contatto con organi vitali.
ARTICOLO 9 D. Lgs 46/97 Informazioni riguardanti incidenti verificatisi dopo l’immissione in commercio Gli operatori sanitari pubblici e privati devono comunicare i dati relativi agli incidenti che hanno coinvolto un dispositivo medico al Ministero della Sanita’
ARTICOLO 10 D. Lgs. 46/97 Monitoraggio I legali rappresentanti delle strutture sanitarie pubbliche e private e gli operatori sanitari sono tenuti a comunicare immediatamente al Ministero qualsiasi alterazione delle caratteristiche e delle prestazioni di un dispositivo o inadeguatezza nelle istruzioni per l’uso
DECRETO del MINISTERO 15/11/2005 approvazione di modelli di schede di segnalazione di incidenti o di mancati incidenti che coinvolgono dispositivi medici Nel decreto sono indicati gli eventi che debbono essere comunicati al Ministero della Salute • INCIDENTE : si intende la condizione in cui qualsiasi disfunzione o deterioramento delle caratteristiche o delle prestazioni, nonché qualsiasi carenza nell’etichettatura o nell’ istruzioni per l’uso abbiano causato un grave peggioramento dello stato di salute o la morte del paziente o di un utilizzatore. • MANCATO INCIDENTE : si intende la condizione in cui qualsiasi disfunzione o deterioramento delle caratteristiche o delle prestazioni, nonché qualsiasi carenza nell’etichettatura o nell’ istruzioni per l’uso avrebbe potuto causare, se il dispositivo fosse stato utilizzato, un grave peggioramento dello stato di salute o la morte del paziente o di un utilizzatore ……. .
DOVE INVIARE LE SEGNALAZIONI DI INCIDENTE E MANCATO INCIDENTE Ministero del Lavoro, della Salute e delle Politiche Sociali Dispositivi medico diagnostici in vitro Ufficio V Via Giorgio Ribotta, 5 00144 Roma Fax 06. 5994. 3812 Ufficio IV Via Giorgio Ribotta, 5 00144 Roma Fax 06. 5994. 3266 Regione Romagna Servizio Politica del Farmaco V. le Aldo Moro, 21 40127 Bologna Fax 051. 527. 7061 Emilia
TEMPI PER LA SEGNALAZIONE La segnalazione deve avvenire nel rispetto dei seguenti termini temporali: • per gli incidenti : immediatamente e comunque non oltre 10 gg dall’evento; • per i mancati incidenti: entro 30 gg dall’evento.
AVVISO DI SICUREZZA FIELD SAFETY NOTICE ( FSN ) Dal 1° ottobre 2008 la pubblicazione degli AVVISI DI SICUREZZA da parte della Direzione generale Farmaci e Dispositivi sul proprio portale (indirizzo web http. //www. ministerosalute. it/dispositivi/archivio. Avvi si. Dispo. jsp? menu=avvisi&lingua=italiano) è la principale modalità di divulgazione degli avvisi. . che consistono in lettere di informazioni di sicurezza FSN che i fabbricanti inviano agli utilizzatori in caso di ritiro dal mercato o altre azioni, denominate azioni correttive di campo -FSCA secondo la definizione prevista dalla linea guida MEDDEV 2. 12 -1 rev. 5
AVVISO DI SICUREZZA FIELD SAFETY NOTICE ( FSN ) La pubblicazione degli avvisi non deve essere intesa né come unico canale di informazione - divulgazione , né come data-base completo per la ricerca di tutti gli FSN inviati dai fabbricanti che, secondo la normativa vigente, sono tenuti ad informare direttamente tutti i soggetti coinvolti nell’uso dei DM oggetto di FSCA
AZIONE CORRETTIVA DI CAMPO FIELD SAFETY CORRECTIVE ACTION (FSCA ) Misura intrapresa dal fabbricante per ridurre il rischio di morte o di grave peggioramento dello stato di salute, legato all’utilizzo di un DM già commercializzato. Viene segnalato tramite un FSN
RECLAMI Le segnalazioni che non presentano le caratteristiche dell’incidente o mancato incidente devono originare invece un reclamo al fornitore. Esempi ( Linea guida MEDDEV 2. 12 -1 rev. 5 ): • inadeguatezza di un DM riscontrata prima dell’uso • evento causato dalla condizioni cliniche del paziente • superamento della data limite d’ utilizzo o della data di scadenza …. .
RUOLO DEL RESPONSABILE AZIENDALE DELLA VIGILANZA • formazione e sensibilizzazione in tema di vigilanza verso il personale sanitario che utilizza i DM oltre che lo sviluppo delle modalità per fornire un’informazione di ritorno al segnalatore • gestione delle schede di segnalazione di incidenti/mancato incidente (centralizzazione delle informazioni, archivio locale, …. ); • gestione dei percorsi per la diffusione degli FSN all’interno dell’Azienda Sanitaria curando il rapporto con gli utilizzatori; • la gestione dei percorsi per gli eventuali ritiri dei DM presenti in reparto o al domicilio dell’assistito; nonché dei medesimi nei rapporti con i Fabbricanti e/o i distributori. , per gli ambiti di competenza.
AUSL DI RAVENNA Organizzazione n. 3 ospedali : Ravenna, Lugo, Faenza per un totale di 1. 058 posti letto di degenza ordinaria e 134 posti letto di degenza DH Dipartimenti (12 ) Medico Internistico I , Medico Internistico II, Chirurgico, Chirurgie Specialistiche, Malattie Digestive e Metaboliche Nefro - urologico , Maternità, infanzia ed età evolutiva, Oncoematologico, Immagini, Patologia clinica e Medicina Trasfusionale, Urgenza – emergenza e Cardiovascolare
ATTIVITA’ DEL FARMACISTA REFERENTE DELLA VIGILANZA ( RAV ) • Assicura il rispetto dell’iter procedurale aziendale in tema di vigilanza • Collabora con gli operatori sanitari alla compilazione della scheda di segnalazione di incidente/mancato incidente assicurandone completezza ed inoltro al Ministero • Adotta le opportune misure per la conservazione dei dispositivi medici oggetto della segnalazione secondo le modalità più idonee • Verifica la presenza di altri dispositivi del medesimo lotto. • Trasmette le comunicazioni ministeriali di ritiro e gli alert delle ditte alle UU. OO. interessate • Mantiene un archivio cartaceo ed informatico relativo alle segnalazioni ricevute
Farmacia Ospedaliera AUSL Ravenna Mdir 1 13 Reclamo / Segnalazione Rev. 1 del 12/09/2000 N° ___ Gentile Utente: il presente modulo Le permette di comunicarci le carenze riscontrate durante la fruizione dei ns. servizi o nei prodotti distribuiti oppure di indicarci i Suoi suggerimenti. Esamineremo quanto da Lei segnalato al fine di trarre indicazioni per un continuo miglioramento. RAVENNA Relativamente alle attività della Farmacia Ospedaliera di Si inoltra il seguente RICHIESTA Il giorno Si è rilevato quanto segue: RECLAMO LUGO FAENZA SEGNALAZIONE / alle ore Ritardo nella consegna Errore nella consegna Articoli mancanti rispetto a quanto indicato in bolla Prodotto difettoso Altro [Solo per Reclamo] a fronte di quanto lamentato si propone la seguente azione di miglioramento: Nominativo del Segnalante Unità Operativa Codice U. O. [Riservato alla Farmacia Ospedaliera] - Esame del Reclamo / Segnalazione / Richiesta FIRMA Modulo NC N° Az. Correttiva N° Az. Preventiva N° Responsabile Qualità Data
Dalle linee guida regionali “SEGNALAZIONE DI RECLAMO” al fabbricante o mandatario o distributore da parte degli operatori sanitari Rapporto interno n. ………. . A) Struttura presso la quale si è verificato l'episodio 1. Denominazione 2. Data dell'episodio 3. Persona di riferimento (o se già presente, il responsabile della vigilanza) 4. Telefono 5. Fax 6. E-mail 7. Struttura sanitaria (ASL, AO) competente per territorio (utilizzare la denominazione ufficiale della struttura)
Dati relativi al dispositivo medico …. Fabbricante (nome, ragione sociale e indirizzo) Mandatario (nome, ragione sociale e indirizzo) (se disponibile) Responsabile dell'immissione in commercio (nome, ragione sociale e indirizzo) (se disponibile) Distributore (nome, ragione sociale e indirizzo) Nome commerciale ed eventuale modello del dispositivo assegnato dal fabbricante Descrizione del dispositivo medico Codice Classificazione unica nazionale dispositivi medici (CND) (quando disponibile nel sito web del Ministero della salute) Numero progressivo di registrazione del dispositivo presso il Ministero della Salute (quando disponibile)
DISPOSITIVO VIGILANZA Attività dal 1 gennaio al 16 novembre 2009 nell’Ausl di Ravenna • Reclami : n. 33 • Avvisi di sicurezza : n. 20 di cui 7 con ritiro di tutti i prodotti coinvolti • Segnalazioni di incidenti : n. 5
ATTIVITA’ AZIENDALI DEL RAV IN PROGRAMMA PER IL 2010 • stesura di una istruzione operativa aziendale in tema di vigilanza sui DM con i servizi coinvolti (Approvvigionamenti, Ingegneria clinica ) • n. 4 incontri di formazione sulla vigilanza per gli operatori sanitari ( medici e personale infermieristico dei diversi dipartimenti) • sviluppo del sistema informatico locale (INTRANET) per una tempestiva e capillare diffusione delle informazioni