2017 MiniBioman Drug Product Manufacturing Day 1 Overview

2017 Mini-Bioman Drug Product Manufacturing Day 1. Overview of parenteral drug products and manufacturing requirements Ivy Tech Community College Bloomington Sengyong Lee, Ph. D Professor and Chair of Biotechnology
 vs Biologics (recombinant proteins etc.")
Types of Pharmaceutical Products • Small molecules (synthetic chemicals) vs Biologics (recombinant proteins etc. ) • Enteral (oral administration) vs Parenteral (intravenous administration) • Generics vs Brand names • Prescription vs Over-the-counter • Blockbuster drugs ($1 billion revenue) vs Orphan drugs (< 200, 000 patients)
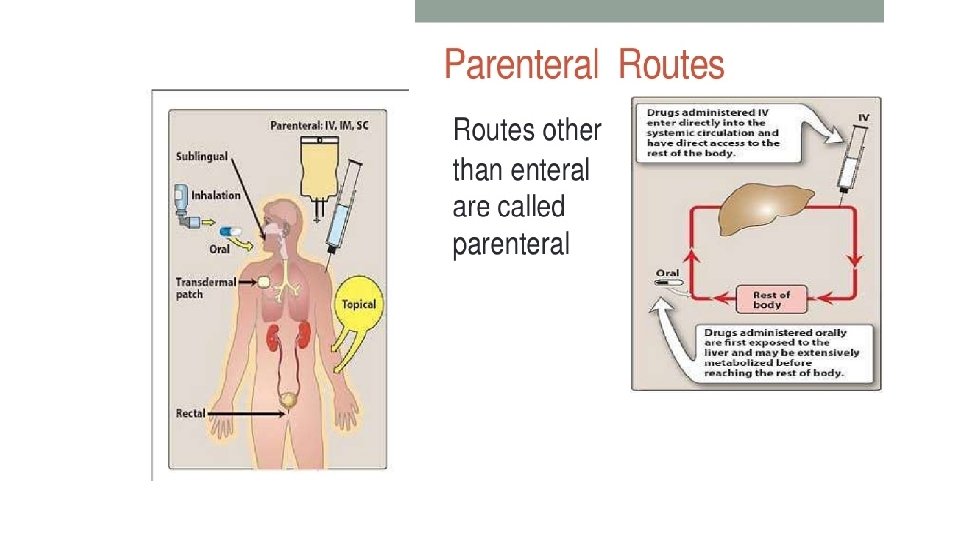

Common Routes of Parenteral Drug Administration
 “enteral” (alimentary canal) meaning")
Parenteral Drugs • “Parenteral” comes from two Greek words, “par”(avoid) “enteral” (alimentary canal) meaning “beside the intestine”. • Pharmaceutical products such as peptides, proteins, and chemotherapeutic agents that can be administered only by injection because if they are given by mouth, they are inactivated in the gastrointestinal tract. • Mostly made up with biological molecules such as human insulin (the first recombinant DNA based protein drug FDA approved in 1982) • Sterile drugs (parenteral + ophthalmic and wound healing products): a product introduced in a manner that circumvents the body’s most protective barriers, the skin and mucous membranes, therefore, must be “essentially free” of biological contamination. • Therefore, sterile drugs are aseptically manufactured and should be free from physical, chemical, and biological contaminants. This is why they have to be manufactured under the current good manufacturing practices (c. GMP).

Drug Product Development • Drug product development is a lengthy, risky, and expensive process. It takes average $1. 1 billion and 12. 5 years to bring a single drug from concept to commercialization. • In the US, the term of the patent is only 20 years from the filing date of its application. GLP/GMP (Pilot Scale) GMP/scale up to Commercial Scale

• Analytical methods are developed prior to the final dosage form, which helps validation efforts prior to IND filing. However, the final assay validation happens after the final formulation is determined. • Clinical phases may take many years with Phase III being the longest. Phase III involves a very large number of subjects spread over several clinical sites to provide statistical verification. • The final formulation, package, and process should be locked in before or during Phase III. • Final production specifications are finalized during Phase III batch production prior to the submission of NDA. • Process validation batch production should be initiated prior to the submission of NDA. Validation report does not have to be completed by NDA but the initial validation data should be available.
 ØDownstream Process")
Manufacturing of Biologic Drugs • Drug Substance Manufacturing ØUpstream Process (cell culture) ØDownstream Process (purification) • Drug Product Manufacturing ØFormulation, filling, lyophilization, capping, packaging, etc. Please review the information on biologics and their manufacturing requirements from the website below. http: //www. gene. com/topics/manufacturing

History of Parenteral Drugs • The first experimental injection was done by Christopher Wren on dogs with crude hypodermic needles in 1656. These experiments consisted of using animal bladders (as the syringe) and goose quills (as the needle) to administer drugs such as opium intravenously to dogs. Their main interest was to learn if medicines traditionally administered orally would be effective intravenously. • In the 1660 s, J. D. Major of Kiel and J. S. Elsholtz of Berlin were the first to experiment with injections in humans. The drug injected was opium for pain but caused microbial and pyrogenic contamination eventually killed the patient. • In 1796, E. Jenner used intradermal injections of cowpox virus to inoculate children against small pox.
 therapy of salt, bicarbonate and water")
• In 1831, the first intravenous (IV) therapy of salt, bicarbonate and water was applied to treat cholera patients. • In the mid-1850 s, the first “hypodermic (under the skin)” needle syringe was used for “subcutaneous” injection. • In the 1860 s, the germ theory (some diseases are caused by microorganisms) was discovered by Pasteur, Lister, and Koch. • In 1884 the first use of autoclave for sterilization was introduced by Charles Chamberland. • Gaseous sterilization was discovered using formaldehyde in 1859 and ethylene oxide in 1944. • Radiation with ultraviolet light was used as a means of sterilization. • Filtration methods began in the mid 1850 s.

• In 1923, Florence Siebert discovered the cause of pyrogenic reaction. She suggested the reaction is caused by something microbial origin, but nonliving, nonprotein based and could not be eliminated by sterilization methods. She also developed the rabbit pyrogen test.

• In 1938, the Food, Drug, and Cosmetic Act was passed by the Congress after the sulfanilamide incident where 107 people died due to ingesting this drug dissolved in diethlyene glycol. This Act established FDA which enforced this Act so manufacturers should prove to the government that drug they produce were safe. This provided the legal basis for c. GMP and other FDA regulations. • In 1940 s, penicillin started being used. • Freeze-drying introduced during WW II

• After the thalidomide incident in Europe, Congress passed the Kefauver-Harris Drug Amendments to the Federal FD&C Act in October 1962. Before marketing a drug, firms had to prove not only the safety, but also the substantial evidence of effectiveness for the product's intended use. The evidence had to consist of adequate and well-controlled studies.

• In 1961, clean room technology, including the use of laminar air flow units, high efficient particulate air (HEPA) filters, was introduced. The first clean room classifications was proposed by the US government in 1962.

• In 1963, FDA’s 21 CFR Parts 210 and 211 under section 501 (a)(2)(B) (https: //www. accessdata. fda. gov/scripts/cdrh/cfdocs/cf. CFR/CFRSearch. cfm ? CFRPart=210&show. FR=1), the first proposed c. GMP regulations was published. A revised and expanded version was published in 1978. ØCFR - The Code of Federal Regulations (CFR) is the general and permanent rules published in the Federal Register by the executive departments and agencies of the Federal Government. The CFR is divided into 50 titles representing broad areas subject to federal regulation. ØEach title is divided into chapters that are assigned to agencies issuing regulations pertaining to that broad subject area. Each chapter is divided into parts and each part is then divided into sections -- the basic unit of the CFR. ØThe purpose of the CFR is to present the official and complete text of agency regulations in one organized publication and to provide a comprehensive and convenient reference for all those who may need to know the text of general and permanent Federal regulations.

• Emergence of biotechnology in the 1970 s. The first biotech drug, humulin, was approved by FDA in the early in 1980 s. (http: //www. fda. gov/About. FDA/What. We. Do/History/Product. Regulation/Selectio ns. From. FDLIUpdate. Serieson. FDAHistory/ucm 081964. htm) • FDA published the Guidelines for Aseptic Processing and Process Validation. • In 1997 the first human monoclonal antibody drug, Rituxan, for cancer was approved by FDA.
 • Since")
Characteristics of Parenteral Drug Forms 1. Safety (freedom from adverse toxicological concerns) • Since parenteral drugs are directly injected into the body, they avoid the body’s natural barriers. Therefore, all the components of the drug should be proven to be safe at the quantity level it is injected. • When selecting excipients to overcome drug’s solubility, stability, tonicity, and delivery, a formulation scientist should carefully follow the safety requirements. • Under the Kefauver-Harris Amendments to the Federal Food, Drug, and Cosmetic Act, pharmaceutical preparations should be tested for safety in animals.
 • Many factors contribute to achieving and maintaining")
2. Sterility (freedom from microbial contamination) • Many factors contribute to achieving and maintaining sterility such as valid sterile procedure for all components of manufacturing, valid procedure for aseptic filtration, design and maintenance of clean rooms, validation of aseptic processes, training and application of good aseptic practices, use of antimicrobial preservatives for multi-dose products, valid testing for sterility of the product and maintenance of container/closure integrity. 3. Nonpyrogenic (freedom from endotoxin contamination) • Pyrogens are fever-inducing entities originating from microbial causing a variety of complications in the human body. • Limulus Amebocyte Lysate (LAL) test is used for the quantitative detection of the bacterial endotoxins in all parenteral drug products. • Various depyrogenation methods include cleaning validation, time limitations, validated depyrogenation cycles for glassware, validation of pyrogen/endotoxin removal from rubber closures and other items based on rinsing techniques, validated water systems, and use of endotoxin free raw materials

LAL Assay
 • Visible particulate matter implicates product quality")
4. Particle-free (freedom from visible particle contamination) • Visible particulate matter implicates product quality and safety. Ready to use and reconstituted solutions should be free from any visible particulate matter and must meet compendial specifications for numbers of subvisible particles no greater or equal to 10 or 25 micrometer. • The factors contribute to the presence or absence of particulate matter include valid cleaning methods of all equipment and packaging materials, valid solution filtration procedures, adequate control of production and testing environments, adequate training of personnel, testing and using sterile product solutions and employment of required compendial testing procedures for detection of both visible and subvisible particulate matter.
 • Parenteral drug products should be stable under")
5. Stability (chemical, physical, and microbial) • Parenteral drug products should be stable under predetermined manufacturing, packaging, storage, and usage conditions. The chemical and physical stability should be maintained throughout the shelf-life of the product. • Complicated chemical structures and vulnerabilities to environmental conditions (temperature, light, p. H, shear, metal impurities, oxygen, etc. ), therapeutic peptides and proteins offer enormous challenges. • Compounding, mixing, filtering, filling, stoppering, sealing, storing, shipping, and handling affect the stability of the product. • Many parenteral drugs are unstable in solution, which challenges their Lyophilization processes further. • Maintenance of sterility throughout the shelf-life and usage also affects the stability of the drug.
 • Many parenteral drugs are manipulated prior")
6. Compatibility (formulation, packaging, and other diluents) • Many parenteral drugs are manipulated prior to injection. Lyophilized products should be reconstituted by sterile dilution, and often combined with another solution or large volume infusion fluid which may contain more than one drug already. This means the parenteral drug must be proven to be compatible with other drugs may be administered together.
 • Biological cells maintain a certain concentration of")
7. Isotonicity (isotonic with biological fluid) • Biological cells maintain a certain concentration of ions , molecules, and other aggregated species that give cells specific properties. Due to such properties osmotic pressure is maintained across the semipermeable cell membrane. • If a parenteral drug that is hypotonic to the biological solution of the cells, the solvent from the injection can move into the cells can cause cell burst (hemolysis of RBCs). If a parenteral drug is hypertonic, an injection could cause cells to shrink, crenation. • Ideally, any injection formulation should be isotonic with biological cells to avoid any potential problems.

• Large volume of IV injection and small volume injections of all kinds other than IV must be isotonic to avoid pain, tissue irritation, and physiological reactions. • Small volume of IV injections do not absolutely have to be isotonic as they do not cause extensive damage to RBCs. • 0. 9% Na. Cl (ionic) solution and 5% dextrose (nonionic) solution are isotonic with biological cells.
 injectables 2. Large volume")
Types of Parenteral Drug Forms 1. Small volume (<100 ml) injectables 2. Large volume (>100 ml) injectables 3. Modified release (sustained-release) injectables (SVIs delivered by routes other than IV)

1. Small Volume Injectables • SVIs include solutions, suspensions, emulsions, and solids. • Most of SVIs are ready to use solution forms and the next frequent type of forms are the lyophilized or powder filled dosage forms. The third forms are suspensions, emulsions, and other dispersed systems. ØSolutions – ready to use products or can be liquid concentrates to be diluted in a small container or in a IV fluid. Aqueous solutions are water based or combined with water miscible organic solvents such as ethanol, polyethylene glycol, glycerin or propylene glycol. Nonaqueous solutions contain vegetable oils such as soybean, sesame, and cottonseed as the vehicle. Oil solutions must be administered by the IV.
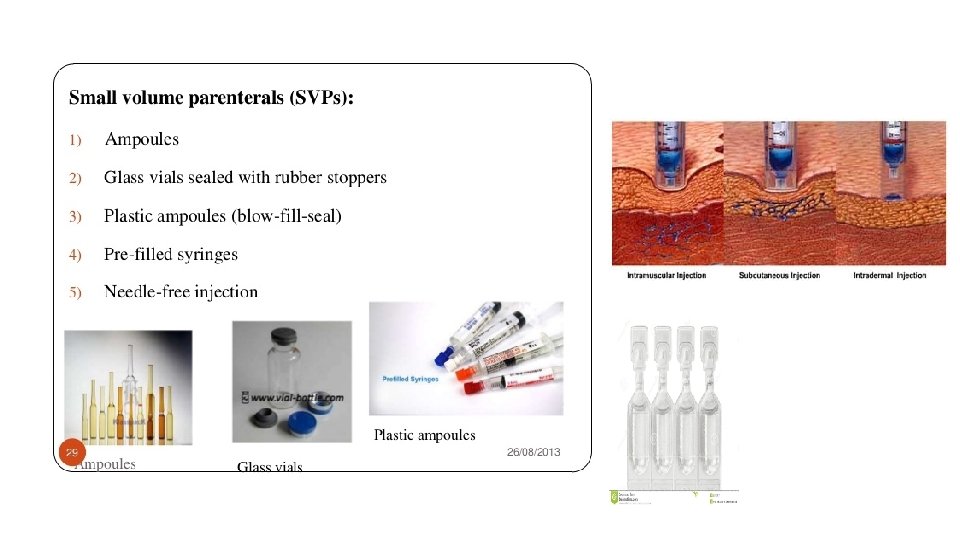

ØSuspensions – macro- or micro-sized solids dispersed in a suitable vehicle, water or oil. Insulin, vaccines, and microsphere are delivered as suspensions. Suspensions are not administered by the IV.

ØEmulsions – dispersed systems combined with an oil phase and an aqueous phase. It could be either oil-in-water emulsion or water-in-oil emulsion, but most injectable emulsions are oil-in-water emulsions. Liposomes are emulsified spherical vesicles composed of a phospholipid bilayer with an aqueous inner phase. Drug can be incorporated in either the liquid or aqueous phases. Parenteral emulsions are milky white in appearance and have an average globule size of 1 – 5 micrometer. Globules larger than 5 micrometer should be less than 0. 05% of the total volume. Propofol and oil soluble vitamins are examples.

ØSolids – primarily prepared by lyophilization as amorphous solids are very difficult to fill accurately due to their lack of density. Some solid formulation that can be crystalized and powder filled. Sterile solids are reconstituted prior to injection with a suitable diluent.

2. Large Volume Injectables Ø LVIs include electrolytes, carbohydrates, proteins, fatty emulsions, dialysis solutions, and irrigating solutions. Ø Electrolyte solutions – 0. 9% Na. Cl isotonic solutions, 0. 45% or 3% Na. Cl, 20 -40 m. Eq/L KCl, Ringer’s lactated Ringer’s, etc. Ø Carbohydrate solutions – Dextrose 5% in water (D 5 W) Ø Nutritional proteins: 2. 5 – 10% synthetic amino acid mixtures used on patients with starvation, renal, hepatic failure or other conditions. Ø Fatty Emulsions: serve as a source of nutrient fat. Composed of 10 -20% soybean oil, water and 1. 2% egg yolk phospholipids, and 2. 5% glycerin. Ø Peritoneal Dialysis: dialysis solution contains large volumes of dextrose to remove waste such as urea and potassium from the blood.
 solutions – they come in various formulations and differ from injectable solutions")
ØIrrigating (washing) solutions – they come in various formulations and differ from injectable solutions with the package closure. Irrigating solutions have a screw cap that twisted open. Irrigating solutions must be sterile, pyrogen and particulate free.

3. Sustained Release Injectables ØSustained or controlled release injectable systems are desirable for increased duration of release, reduced number of injections, and increased compliance, localized delivery in the case of cancer therapy and vaccinations, and protection against in vivo degradation of the active ingredient.

ØPolymeric implants – sterile, solid drug products manufactured by compression, melting or sintering processes. They have drug and a biodegradable or replaceable polymeric system which controls the sustained or prolonged drug delivery. Norplant (birth control implant) is shown in the picture below.

ØMicrospheres: injectable suspensions containing particles of diameters of 1 – 100 micrometer. Prior to injection, the particles are mixed with an appropriate vehicle, dispersed and administered. Drug release kinetics are controlled by polymer degradation and diffusion. ØLiposomes - a spherical vehicle having at least one lipid bilayer. The liposome can be used as a vehicle for administration of nutrients and drugs.
 process")
Overview of Parenteral Drug Manufacturing • The parenteral drug manufacturing (Drug Product Manufacturing) process includes compounding, mixing, filtration, filling, terminal sterilization, lyophilization, closing, and sealing, sorting, and inspection, labeling, and final packaging for distribution. • The manufacturing process is complicated, requiring organization and control to ensure the product meets the quality and the specifications. • Aseptic processing requirement adds more complication but assures that all dosage forms manufactured are free from any contamination of microbial, endotoxin, and visible particulate matter. • The manufacturing process initiates with the procurement of approved raw materials (drug, excipients, vehicles, etc. ) and primary packaging materials (containers, closures, etc. ) and ends with the sterile product sealed in its dispensing package.
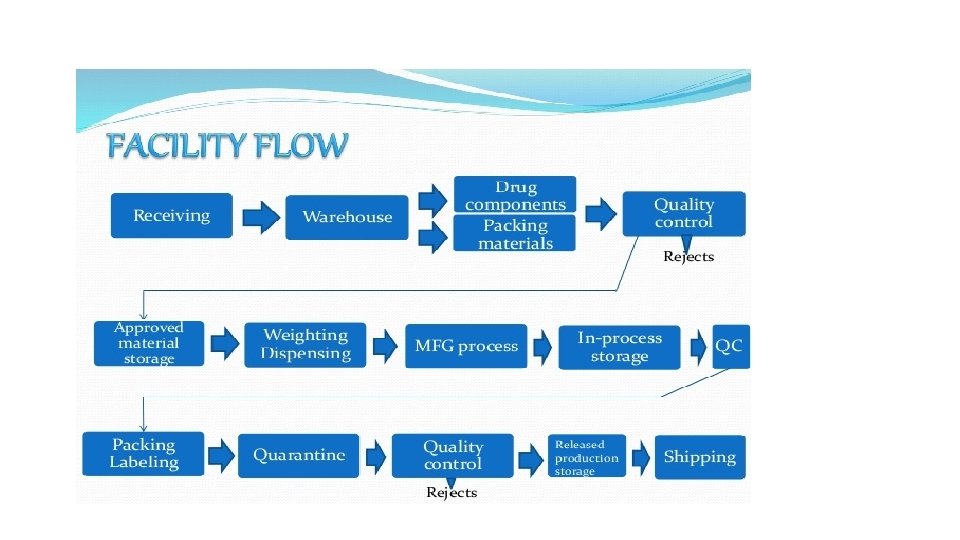

• To ensure the product meets required quality, each manufacturing process should be validated to be sure that it is accomplishing what it is intended to do. For example, any sterilization process should be validated with data showing it is killing various forms of microorganisms. • The validation processes are integral part of c. GMP. • Small scale dispensing (product batch size in hundreds) – for early phase of clinical trial • Large scale dispensing (product batch size in hundreds of thousands) - for late phase of clinical trial or commercialization

c. GMP Principles and Compliance Title 21 CFR Part 211 • CFR 211. 25 (https: //www. accessdata. fda. gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch. cfm? fr=21 1. 25) – personnel qualification requirement based on education, experience and training for the job. • CFR 211. 80, 211. 84, 211. 86 – ingredients used in product compounding meet the required identity, quality, and purity • CFR 211. 63, 211. 65, 211. 67, 211. 68, 211. 182 – validation requirement for processes and equipment used • CFR 211. 42, 211. 46, 211. 56, 211. 113 – requirements for the production environment preventing any contamination • CFR 211. 160, 211. 165, 211. 167 – requirements for quality control procedures ensuring the finished products have the required potency, purity, and quality until the expiration date • and so forth ---

c. GMP Principles and Compliance

Batch record and other documentation • Batch record is the most important document in pharmaceutical product manufacturing as it is the complete record of the manufacturing control, distribution of a single patch of a product. • Batch record should be accurate, complete, readily followed, and reviewed. • Batch record should contain: product identification, document identification, company name, dates of manufacturing, step-by-step list of unit operations and tests, specifications/limits of each step, formulation information (names, quantities, ID numbers), control numbers for each component with quality approval, start and completion time for each operation, chemical weight check (QA counter check), ID of all processing equipment, process details, deviation investigations/reports, labeling requirements, in process sampling procedures, test requirements, final test results, material accountability, signatures.
 are activities require that all raw data, written")
• Good documentation practices (GDP) are activities require that all raw data, written entries, and records to be accurate, legible, traceable (defendable), reproducible, complete, and verified. • All entries must be signed and dated by the individual who made the entry on the date the entry was made. • All record must contain proper identification on each page. • When mistakes are made, never erase a mistake, add single line cross outs, add correct entry with one’s initials, date, and reason for the correction. • Documentation failures are the most frequently cited observations during FDA and other inspections. • Electronic batch record should also substantiate that every significant step in the production and packaging was accomplished according to GMP.

Manufacturing Facilities • The manufacturing facilities should be designed, constructed, and operated properly for the production of a pharmaceutical product with the excellent quality required for safety and effectiveness. • Materials used in manufacturing facility must be smooth, cleanable, and impervious to moisture and other damage. • All connections or junctions between ceilings, walls, and floors cannot be 90 degree angles but covered for easy cleaning. • There should be no gaps, cracks, recesses, or other defects that can harbors for microbial contamination. • Air systems, lighting, and communication systems require special design to be easily accessible without disturbing the normal air flow. • Air pressure should be controlled so air flows from higher classifications (Grade A) to lower classifications (Grade C). • Doors should be self-closing and interlocked so only one door opens at a time.

c. GMP regulation requirements on facility: • Section 211. 42: There must be separate or defined areas of operation to prevent contamination, and that for aseptic processing there be, as appropriate, an air supply filtered through HEPA filters under positive pressure, and systems for monitoring the environment and maintaining equipment used to control aseptic conditions. • Section 211. 46: Equipment for adequate control over air pressure, microorganisms, dust, humidity, and temperature be provided where appropriate, and that air filtration systems including prefilters and particulate matter air filters, be used when appropriate on air supplies to production areas.

Clean room classified areas • Manufacturing areas have standard criteria for the acceptable maximum limit of airborne particles greater than or equal to 0. 5 micron per cubic foot or meter. • Air Classification Systems – accordance with the FDA and EU guidelines • Particle counts must be measured during filling and closing operations.

• There are three clean room standards: Federal, ISO and EU


• Grade A/B or Class 100 clean room must have 90 -100 ft/min+20% airflow determined by smoke tests (using glycerol based fog generator). • All HEPA filters should be integrity tested twice a year using poly-alpha-olefin (PAO) aerosol. This aerosol is polydispersed, nontoxic liquid that possessed a light scattering droplet with 0. 7 micrometers. Inside of the barrier/isolator Inside of the fill-in room where operators are

• Flow Plan – the key in production facility design is to ensure that movement of equipment, materials, and people is unidirectional, eliminating any crossover of clean and dirty equipment.

Barrier/ isolator Technologies • Barriers/ isolators provide separation between the external clean room and a work process. Isolator design is more dependable than barrier design. • Barriers/ isolators are ideal for smaller facilities and much more economical from the point of labor and maintenance, and operator costs. • Barriers/ isolators can protect the product from human contamination as well as protect the human from potential toxic effect of direct exposure to the drug product (cytotoxic drugs).

• Isolator – provide complete isolation of the inside of isolator (Class 100/ISO 5/Grade A) from external environment (Class 10, 000/ISO 7/Grade B) • Restricted Access Barrier Systems (RABS) – provide sub-isolation protection but far better than the laminar flow hoods. Passive RABS has its air provided by the facility (clean room) while an active RABS has its own HVAC and HEPA system. • RABS and isolator systems use glove ports and rapid transfer ports (RTPs).

• Disinfection is typically manual in a standard RABS but there is no operatorhuman access to the isolators as their product contact parts are cleaned and sterilized in place (CIP/SIP). • Validation of barrier isolation Ø Air supply specification – air change rate, air velocity, particulate air specification, recirculation rate, temperature and humidity, aeration of the decontaminating agent Ø Leak testing – pressure decay test, tracer gas detection test Ø Rapid transfer ports – seal integrity Ø Facility requirements – classification of isolator room, temperature and humidity control, process utilities Ø User requirements – sterilization and decontamination methods, cleaning, contaminant, environment control and monitoring, Microbiological monitoring, process simulation

• Operation qualification – all alerts and alarms should be tripped and verified, integrity check should be verified to be free from leaks, sterilization cycle verification should include temperature and humidity control as well as the concentration and distribution of the sterilization gas during the sterilization cycle. • Sterilization cycle development – sterilization should be demonstrated by use of biological indicators (BI) such as Bacillus stearothermophilus. BI kills of 103 -106 are commonly used.

Water and Air Quality • Water is the most commonly used solvent in the pharmaceutical production. It is used for: Ø Ø Ø Solvent in formulation Cleaning of components and equipment Solvent in cleaning, sanitizing, disinfectant solutions Source of clean steam Source of cooling water for freeze-dryer compressors Source of water for chillers needed for air compressor, rubber closure processors, cooling of depyrogenation tunnels • United States Pharmacopeia (USP) and other compendial specifications for the water used in pharmaceutical manufacturing, Water for Injection (WFI) limits the amount of endotoxins (0. 25 EU/ml).
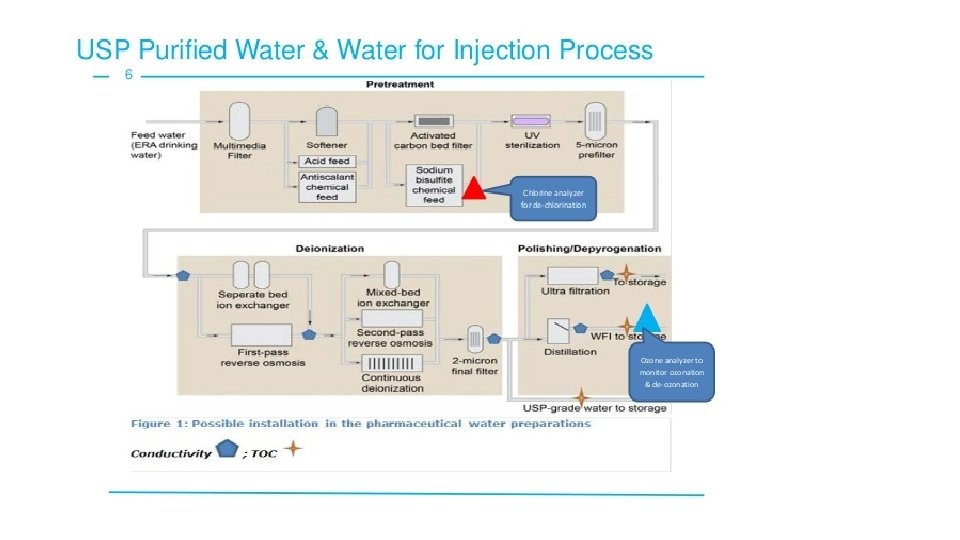
 or Ultrafiltration")
• WFI can be prepared by distillation or Reverse Osmosis (RO) or Ultrafiltration (UF). The European Pharmacopeia (EP) permits distillation as the only process for producing WFI, but the USP and Japanese Pharmacopeia (JP) allow other technologies as well. • Potential impurities in feed water (city water) include bacteria, endotoxins, particles, electrolytes, organics, colloids, and disinfectants such as chlorine. • Water purification methods include: Ø Distillation – a process of converting water from a liquid to its gaseous form. Since steam is pure and other contaminants are removed.
 – pressure, 200 -400")
• Water purification methods include: Ø Reverse Osmosis (RO) – pressure, 200 -400 psig is applied to force pure water to permeate through a semipermeable membrane that filters contaminants.

• WFI is either collected in a holding tank or recirculated through facility piping systems. The holding tank with several thousand gallon capacity is normally kept at a constant 80 degree in Celsius to prevent microbial growth (the requirement by the USP). If stored at room temperature, any unused WFI should be discarded after 24 hours. • USP and EP provide the official standards of purity for WFI. Ø Ø Inorganic content – water conductivity at 25 o. C, <1. 3 m. S/cm Organic content – total organic carbon, <0. 5 mg/L Pyrogen content – LAL test, <0. 25 EU/ml Microbial content – total bacterial count, <10 CFU/100 ml

• Air is one of the greatest potential source of contamination in clean room, a series of treatments and monitoring systems should be in place. • Air from the outside is passed through a prefilter (glass wool, cloth, or shredded plastic) that removes large particles and then, it is treated with an electrostatic precipitator that removes charged particles. The air then passes through the HEPA filter. • HEPA filter can remove 99. 97% of particles >0. 3 mm. It is made with densely packed fiberglass.
 Preparation of")
• The air classification of sterile production area: Ø Warehouse (Unclassified) Preparation of equipment /components (Class 100, 000) Compounding of the product (Class 100, 000) Filling (Class 100) Capping (Class 100, 000) Sterilization (Unclassified) Sampling (Unclassified) Finishing (Unclassified)

Contamination Control • Therapeutic protein products are natural nutrient sources for microorganisms and are typically formulated at neutral p. H and isotonic that are ideal for microbial growth. • There have been so many product recalls initiated by FDA due to suspected microbial contamination of products. • Sources of microbial Contamination includes: ØThe atmosphere – certain microorganisms like Bacillus, Clostridium, Staphylococcus, Streptococcus, and Penicillium can tolerate dry environment and can be the source of contamination. ØWater – Pseudomonas sp. , Bacillus sp. , and Escherichia coli are indigenous to water and are the main source of contamination. ØRaw materials – many raw materials originate from plants or animals and can be contaminated with pathogens like E. coli and Salmonella. ØPackaging – cardboard container, rubber closures, papers with moisture support the growth of mold spores (Penicillium and Aspergillus) and bacteria (bacillus).

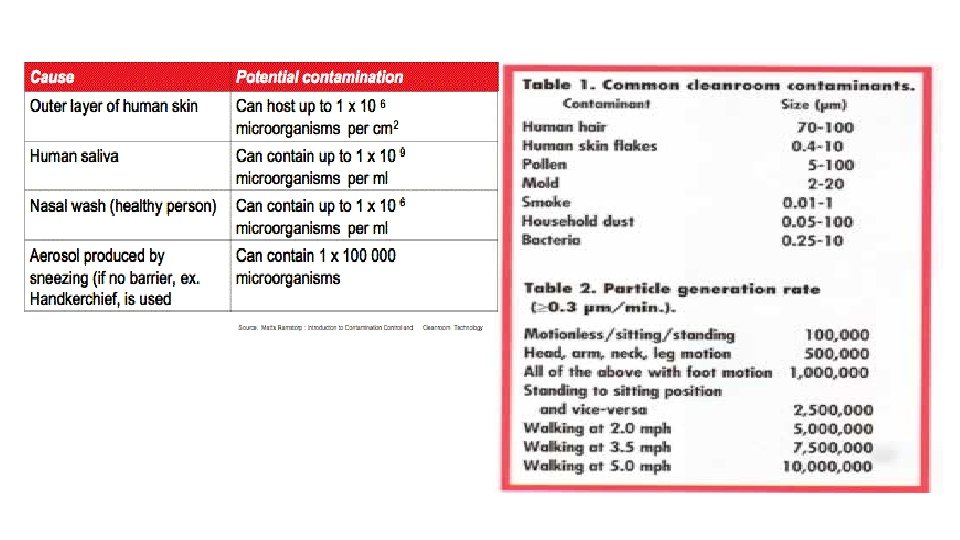
ØBuildings – walls, ceilings, and plasters can contain mold contamination. Floors with cracks filled with water can also offer contamination of microbial growth. ØEquipment – hard to clean locations like screw threads, agitator blades, valves, and pipe joints can harbor microbial contaminations. ØPeople – the largest single source of contamination. Skin, hair, moisture from breathing, coughing, sneezing, cosmetics and clothing are sources of microbial contamination. Movement like a simple walking can emit more than 10, 000 particles per minute.

1. Contamination Control


Clean Rooms & Microbiology – Activities vs. Particles Generated Sitting Quietly Moving Walking Particles shed per minute 100, 000 Horseplay 1 Million 5 Million 100, 000

• Clean room maintenance Ø Carefully planned schedule of cleaning by an expertly trained custodians should be implemented. Cleaning tools should be nonlinting. Ø Sanitization/disinfection agents – chemicals reducing the microbial bioburden. They are not sterilization agents! Ø Quaternary ammonium compounds, phenolic mixtures, alcohols, biguanides, formaldehyde, chloride, peroxide, glutaraldehyde, etc. Ø Lp. H (Steris Corp. ) is well known. Sanitizing agents are normally not sporocides!

 is also an effective sporicidal agent.")
* 5% bleach (sodium hypochlorite) is also an effective sporicidal agent.

• Choices of sanitizing agents must be validated for their effectiveness. A 2 to 3 log reduction (99 -99. 9% killing) in inoculum (typical environmental isolates) should be demonstrated. • Sanitization can be classified as: Ø Deep-cleaning (done after an area shutdown) Ø Routine (daily or some other frequency during normal operation) Ø Continuous (frequent sanitization on gloved hands and utensils used during manufacturing) • Disinfectants should be rotated with sufficient frequency (weekly or monthly) to avoid the development of resistant strains (the European GMP guideline). • Three bucket (sanitization solution, WFI, squeeze drain) sanitizing system is used for facility sanitization.

• Containers and equipment coming in contact with parenteral preparation must be cleaned with extreme care. Equipment should be reserved and used for one product. • Various types of container washers are used for various types of sterile product containers but the basic principles of cleaning should adhere to; Ø The liquid or air should be introduced in such manner that it will strike the bottom of the inside of the inverted container, spread in all directions, and flow down the walls and out the opening with a sweeping action. Ø Cleaning should alternate hot and cool treatments with the final rinse using WFI. Ø Metal components of the washing/ cleaning equipment should be constructed with stainless steel or other noncorroding and noncontaminating material. Ø Detergent are not used for new containers due to the risk of leaving residues. Ø One or two hour treatment at 250 OC is adequate for depyrogenation. Ø Rubber closures are cleaned with gentle agitation in a hot solution of mild water softener or detergent followed by several rinse with WFI. Ø Cleaning equipment should be cleaned in placed (CIP).

Environmental Control Evaluation • Environmental monitoring programs obtain the information on microbial and particulate matter on the following: Ø Ø Ø Room/facility People Utilities such as water, compressed gases, clean steam HEPA filters Filling nozzles Performance quantification studies • The test determines the level of viable and nonviable particles suspended in air or settled on surfaces in the workplace. • If high individual counts, a rising trend, or other abnormalities are reported, corrective action and follow-up measures should be required.


CFUs are determined 48 hours after the incubation at 30 – 35 o. C

• Controls of pyrogens – depyrogenation on glassware and equipment is maintaining a dry heat of 250 o. C for 45 minutes, 650 o. C for one minute, and 180 o. C for four hours. Ø Heating with strong alkali or oxidizing solutions will destroy pyrogens as well. Ø Thorough washing with detergent followed by thorough rinse with pyrogen free water could make glassware and rubber stoppers pyrogen free. Water is the greatest potential source of pyrogen contamination. Ø Anion exchange resins and positively charged membrane filters can remove pyrogen from water. Ø Distillation and RO membranes are also effective depyrogenation treatments.

Personnel Requirements for Sterile Manufacturing • People are the worst source of microbial and particle contamination. • Personnel selected to work in clean room must be neat, orderly, and reliable. They should be in good health and free from dermatological conditions that may increase the microbial load. A person with cold, allergies, or other illness should not be permitted in the cleanroom until their recovery. Effective training and proper gowning can reduce the particle shedding from personnel. • Bathing removes microorganisms but increases the number of particles emitted form human body. Personnel working in cleanroom should bathe at least 2 hours before they enter clean room. • Suntan also dries skin, causing it to flake and peel more easily. Cleanroom personnel are encouraged not to expose their skin to excessive sunlight. • Friction between clothing and skin also increases the rate of shedding. Therefore, tight clothing should be avoided.

• Airborne contamination is directly related to the number of people working in a given area. Number of people and degree of activities must be kept to a minimum. Talking, unnecessary bodily movement and anything that interrupts the flow of laminar air should be avoided. • Cleanroom gowns are designed to confine the contaminants discharged from the body of the operator, preventing the entry into the cleanroom. • Fresh and sterile gowns should be used whenever an individual returns to the aseptic area. • Full gowns consist of coveralls, hoods, face masks, goggles, boot covers, and double gloves. • There is no universally accepted gowning procedure for the sequence of gowning steps but the general sequence is described in the next page.


• Online interactive animation gowning simulation training http: //www. ncbionetwork. org/educational-resources/elearning/interactiveelearning-tools/sterile-gowning-procedures • Gowning video (https: //www. youtube. com/watch? v=uvha. Pu 1 -fd. E)
- Slides: 79