2017 1 Text Harpers Biochemistry Refs Devlins Lippincotts

2017년 1학기 Text : Harper’s Biochemistry Refs: Devlin’s, Lippincott’s AMINO ACIDS METABOLISM Ch. 27. Biosynthesis of the Nutritionally Nonessential Amino Acids Ch. 28. Catabolism of Proteins & of Amino Acid Nitrogen Ch. 29. Catabolism of the Carbon Skeletons of Amino Acids Ch. 30. Conversion of Amino Acids to Specialized Products Carbons NH 3

AMINO ACIDS METABOLISM Carbons NH 3 Urea

Chapter 29. Catabolism of the Carbon Skeletons of Amino Acids

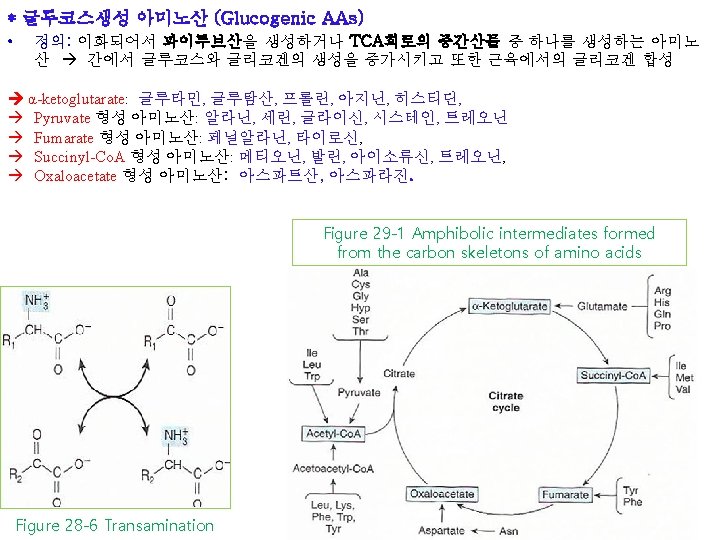
* Transamination typically initiates amino acid catabolism ① Removal of α-amino nitrogen by transamination (Fig. 27 -3, Fig. 28 -6) is first catabolic reaction except for Pro, Hyp, Thr, Lys ② Residual hydrocarbon skeleton amphibolic intermediates (Fig. 29 -1) Figure 29 -1 Amphibolic intermediates formed from the carbon skeletons of amino acids Figure 28 -6 Transamination Alanine (Aspartate) Aminotransferase Figure 27 -3 Formation of alanine by transamination of pyruvate

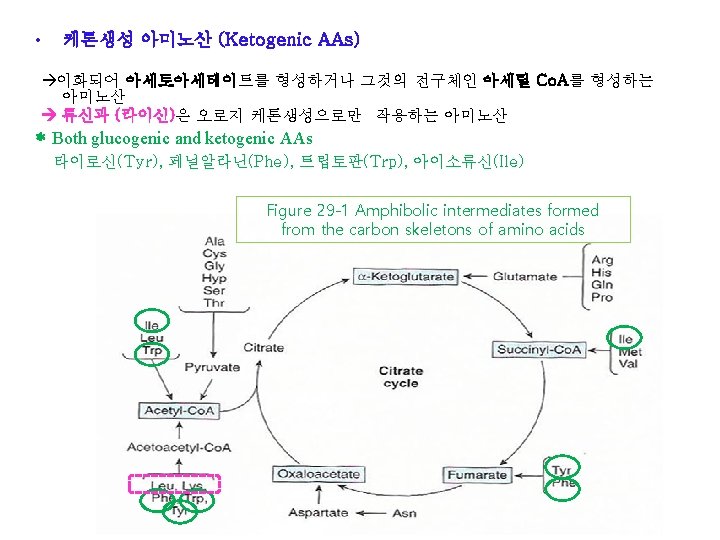
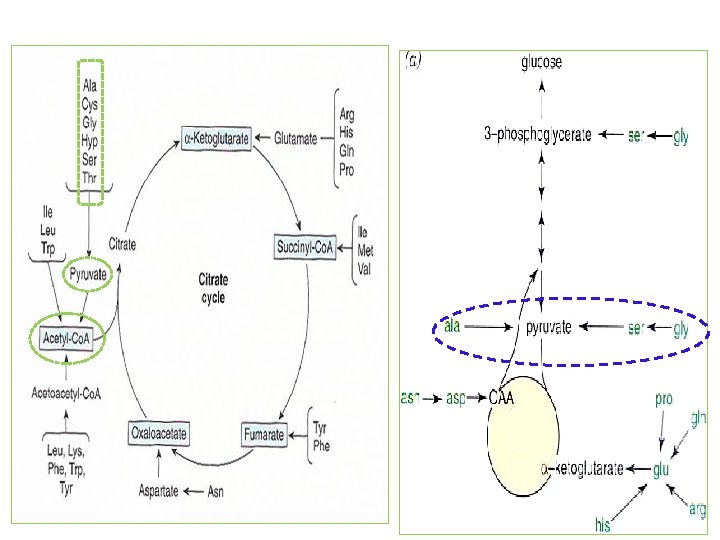
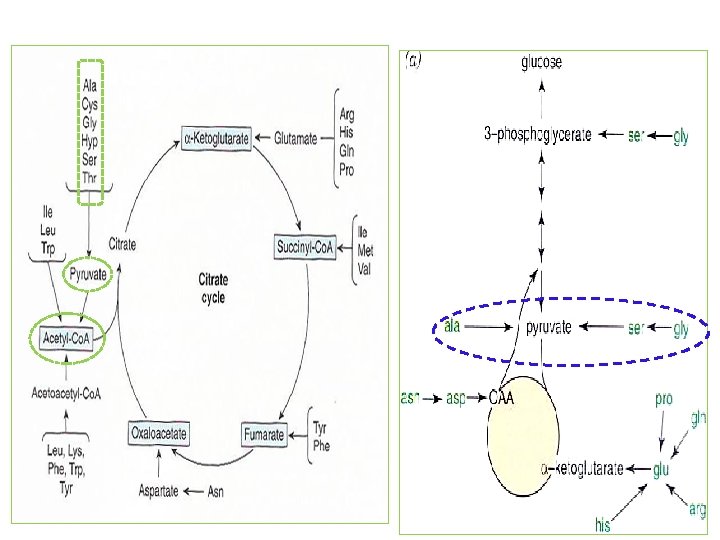
* Transamination typically initiates amino acid catabolism ① Removal of α-amino nitrogen by transamination (Fig. 27 -3, Fig. 28 -6) is first catabolic reaction except for Pro, Hyp, Thr, Lys ② Residual hydrocarbon skeleton amphibolic intermediates (Fig. 29 -1) Glucogenic (글루코스 생성) amino acids Ketogenic (캐톤 생성) amino acids Both glucogenic and ketogenic amino acids Figure 29 -1 Amphibolic intermediates formed from the carbon skeletons of amino acids
 Asparagine, aspartate, glutamine, and glutamate")
• Transamination typically initiates amino acid catabolism 1) Asparagine, aspartate, glutamine, and glutamate ① Carbons of Asn and Asp oxaloacetate ② Carbons of Gln and Glu α-ketoglutarate (Fig. 29 -2) Figure 28 -6 Transamination Figure 29 -1 Amphibolic intermediates formed from the carbon skeletons of amino acids
 Asparagine, aspartate, glutamine, and glutamate Carbons of Asn and Asp oxaloacetate Carbons of")
1) Asparagine, aspartate, glutamine, and glutamate Carbons of Asn and Asp oxaloacetate Carbons of Gln and Glu α-ketoglutarate Figure 29 -2 Catabolism of L-asparagine (top) and of L-glutamine (bottom) to amphibolic intermediates
 Asparagine, aspartate, glutamine, and glutamate Carbons of Asn and Asp oxaloacetate Carbons of")
1) Asparagine, aspartate, glutamine, and glutamate Carbons of Asn and Asp oxaloacetate Carbons of Gln and Glu α-ketoglutarate
 Proline ① Pro dehydroproline glutamate-γ-semialdehyde glutamate α-ketoglutarate (Fig. 29 -3) ② Metabolic block")
2) Proline ① Pro dehydroproline glutamate-γ-semialdehyde glutamate α-ketoglutarate (Fig. 29 -3) ② Metabolic block in type I hyperprolinemia : Pro dehydrogenase ③ Type II hyperprolinemia : glutamate- γ-semialdehyde dehydrogenase type I hyperprolinemia Type II hyperprolinemia Figure 29 -3 Catabolism of proline (mitochondria)

Proline Pro from Glu by reversal of reactions of Pro catabolism Figure 27 -8 Biosynthesis of proline from glutamate
 Proline ① Pro dehydroproline glutamate-γ-semialdehyde glutamate α-ketoglutarate (Fig. 29 -3)")
2) Proline ① Pro dehydroproline glutamate-γ-semialdehyde glutamate α-ketoglutarate (Fig. 29 -3)
 Arginine and ornithine ① Arg ornithine glutamate-γ-semialdehyde")
Figure 29 -4 Catabolism of arginine 3) Arginine and ornithine ① Arg ornithine glutamate-γ-semialdehyde (Fig. 29 -4) Refer to Pro catabolism (Fig. 29 -3) Gyrate atrophy of the retina Figure 29 -3 Catabolism of proline (mitochondria) Glutamate α-ketoglutarate
 Arginine and ornithine ② Hyperornithinemia-hyperammonia syndromes: A defective mito. ornithine-citrulline antiporter (Fig. 28")
3) Arginine and ornithine ② Hyperornithinemia-hyperammonia syndromes: A defective mito. ornithine-citrulline antiporter (Fig. 28 -12) : impaired transport of ornithine into mitochondria for use in urea synthesis Figure 29 -4 Catabolism of arginine Glutamate α-ketoglutarate
 Histidine ① His urocanate 4 -imidazolone-5 -propionate N-forminoglutamate (Figlu) Glu (by formimino group")
4) Histidine ① His urocanate 4 -imidazolone-5 -propionate N-forminoglutamate (Figlu) Glu (by formimino group transfer to tetrahydrofolate) α-ketoglutarate (Fig. 29 -5) ② In folic acid deficiency -> Figlu is excreted ③ Benign disorders: histidinemia, urocanic aciduria associated with impaired histidase Pro. Arg Figure 29 -5 Catabolism of L-histidine to -ketoglutarate

*Catabolism of Glycine, Serine, Alanine, Cysteine, Threonine and 4 -Hydroxyproline ① All of carbons of Gly, Ser, Ala, Cys, Thr, 4 -Hyp form pyruvate acetyl-Co. A Figure 29 -1 Amphibolic intermediates formed from the carbon skeletons of amino acids
 Glycine ① Gly cleavage complex of liver mito: splits Gly to CO 2")
1) Glycine ① Gly cleavage complex of liver mito: splits Gly to CO 2 and NH 4+, and forms N 5, N 10 -methylene THF (Fig. 29 -6) ② Glycinuria : defect in renal tubular reabsorption ③ Defect in primary hyperoxaluria : failure to catabolize glyoxylate formed by deamination of glycine ④ Oxidation of glyoxylate to oxalate urolithiasis, nephrocalcinosis, and early mortality Glycine aminotransferase Dihydrolipoamide dehydrogenase Glycine dehydrogenase (decarboxylating) Glu or Ala ; Amino donor hyperoxaluria Amino-forming aminomethyltransferase Figure 29 -6 The glycine cleavage system of liver mitochondria
 Serine ① Ser to Gly by Ser hydroxymethyltransferase (Fig. 29 -7) ② Ser")
2) Serine ① Ser to Gly by Ser hydroxymethyltransferase (Fig. 29 -7) ② Ser catabolism Gly , pyruvate Figure 29 -7 Interconversion of serine and glycine by serine hydroxymethyltransferase 3) Alanine ① Transamination of Ala pyruvate ② No known metabolic defect Figure 27 -3 Formation of alanine by transamination of pyruvate
 Cysteine ① Cystine to cysteine by cystine reductase (Fig. 29 -8) ② Two")
4) Cysteine ① Cystine to cysteine by cystine reductase (Fig. 29 -8) ② Two different pathways : cysteine to pyruvate (Fig. 29 -9) Figure 29 -8 The cystine reductase reaction Figure 29 -9 Catabolism of L-cysteine via the cysteine sulfinate pathway and by the 3 -mercaptopyruvate pathway
 Cysteine ① Cystine to cysteine by cystine reductase (Fig. 29 -8) ② Two")
4) Cysteine ① Cystine to cysteine by cystine reductase (Fig. 29 -8) ② Two different pathways : cysteine to pyruvate (Fig. 29 -9) ③ Numerous abnormalities of Cys metabolism: Cystine-lysinuria (cystinuria; 시스틴뇨증) (benign); * defects in renal absorption of AAs (Cys, Lys. Arg. Ornithine) * 신장의 근위세관(proximal tubule)에서 시스틴, 라이신, 아지닌, 오니틴을 재흡수하는 체계가 결핍되어 일어나는 질환. 시스틴을 재흡수하지 못하여 신장결석이 생성 mixed disulfide of cysteine and homocysteine excreted by cystinuria patients is more soluble than cystine and reduces formation of cystine calculi (시스틴 결석) (29 -10) Figure 29 -10 Mixed disulfide of cysteine and homocysteine Figure 29 -8 The cystine reductase reaction
 Cysteine ① Cystine to cysteine by cystine reductase (Fig. 29 -8) ② Two")
4) Cysteine ① Cystine to cysteine by cystine reductase (Fig. 29 -8) ② Two different pathways : cysteine to pyruvate (Fig. 29 -9) ③ Numerous abnormalities of Cys metabolism: Cystine-lysinuria (cystinuria; 시스틴뇨증) (benign); mixed disulfide of cysteine and homocysteine (29 -10) Homocystinurias: * deficiency in the reaction catalyzed by cystathionine β-synthase * osteoporosis (골다공증), mental retardation (정신지체) se a h nt Met i e n i on h ts at Figure 27 -9 Conversion of homocysteine and serine to homoserine and cysteine cy sy β
 Cysteine ① Cystine to cysteine by cystine reductase (Fig. 29 -8) ② Two")
4) Cysteine ① Cystine to cysteine by cystine reductase (Fig. 29 -8) ② Two different pathways : cysteine to pyruvate (Fig. 29 -9) ③ Numerous abnormalities of Cys metabolism: Cystine-lysinuria (cystinuria; 시스틴뇨증) (benign); Cystine calculi ; mixed disulfide of cysteine and homocysteine (29 -10) Homocystinurias: * deficiency in the reaction catalyzed by cystathionine β-synthase * osteoporosis, mental retardation Cysinosis (cysteine storage disease): * defective carrier-mediated transport of cystine * deposition of cystine crystals in tissues * early mortality from acute renal failure (급성신부전) Figure 29 -8 The cystine reductase reaction
: Met SAM homocysteine (Fig. 29 -19) L-Homecysteine")
Cysteine Cys from Met (nutritionally essential AA): Met SAM homocysteine (Fig. 29 -19) L-Homecysteine + Ser Cystathionine L-Cys and homoserine (Fig. 27 -9) se a h nt e n i on sy β i cy h ts at Figure 27 -9 Conversion of homocysteine and serine to homoserine and cysteine
 from Met (nutritionally essential AA) : Met SAM homocysteine")
Cysteine Cys (nutritionally nonessential AA) from Met (nutritionally essential AA) : Met SAM homocysteine (Fig. 29 -19) L-Homecysteine + Ser Cystathionine L-Cys and homoserine (Fig. 27 -9) SAM (Fig. 29 -19) Propionyl-Co. A Figure 27 -9 Conversion of homocysteine and serine to homoserine and cysteine
 Threonine ① Thr acetaldehyde + Gly and pyruvate ② Oxidation of acetaldehyde to")
5) Threonine ① Thr acetaldehyde + Gly and pyruvate ② Oxidation of acetaldehyde to acetate formation of acetyl-Co. A (Fig. 29 -11) Ser pyruvate Figure 29 -11 Conversion of threonine to glycine and acetyl-Co. A Figure 29 -7 Interconversion of serine and glycine by serine hydroxymethyltransferase
 4 -Hydroxyproline ① Catabolism of Hyp α-keto- γ-hydroxyglutarate ② An aldol-type cleavage glyoxylate")
6) 4 -Hydroxyproline ① Catabolism of Hyp α-keto- γ-hydroxyglutarate ② An aldol-type cleavage glyoxylate + pyruvate (Fig. 29 -12) ③ hyperhydroxyprolinemia (benign) : A defect in hydroxy. Pro dehydrogenase Type II hyperprolinemia hyperhydroxyprolinemia Figure 29 -12 Intermediates in L-hydroxyproline catabolism
 Tyrosine ① Tyr p-hydroxyphenylpyruvate homogentisate maleylacetoacetate fumarylacetoacetate")
Additional AAs that form acetyl-Co. A 1) Tyrosine ① Tyr p-hydroxyphenylpyruvate homogentisate maleylacetoacetate fumarylacetoacetate fumarate acetoacetate acetyl-Co. A (Fig. 29 -13) Figure 29 -13 Intermediates in tyrosine catabolism

Figure 29 -1 Amphibolic intermediates formed from the carbon skeletons of amino acids
 Tyrosine Type II tyrosinemia (Richner-Hanhart syndrome) (reaction")
Additional AAs that form acetyl-Co. A 1) Tyrosine Type II tyrosinemia (Richner-Hanhart syndrome) (reaction 1) Neonatal tyrosinemia : lower p-hydroxyphenylpyruvate hydroxylase activity (reaction 2) Alkaptonuria (reaction 3) Type I tyrosinemia (tyrosinosis) (reaction 4) Figure 29 -13 Intermediates in tyrosine catabolism
 Tyrosine Alkaptonuria (1859, Garrod’s classic ideas of")
Additional AAs that form acetyl-Co. A 1) Tyrosine Alkaptonuria (1859, Garrod’s classic ideas of heritable metabolic disorder) : lack of homogentisate oxidase : urine darkens due to oxidation of homogentisate : arthritis and connective tissue pigmentation (ochronosis) Alkaptonuria (reaction 3)
 Phenylalanine ① Phe Tyr (Fig 27 -10) subsequent reactions of tyrosine (Fig. 29")
2) Phenylalanine ① Phe Tyr (Fig 27 -10) subsequent reactions of tyrosine (Fig. 29 -13) ② Hyperphenylalaninemias : defects in phenylalanine hydroxylase (Type I, classic phenylketonuria or PKU), or in dihydrobiopterin reductase (Types II and III), or in dihydrobiopterin biosynthesis (types IV and V) ③ Alternative catabolites (Fig. 29 -14) phenylalanine hydroxylase Figure 27 -10 The phenylalanine hydroxylase reaction Figure 29 -14 Alternative pathways of phenylalanine catabolism in phenylketonuria
 Phenylalanine ② Hyperphenylalaninemias : defects in phenylalanine hydroxylase (Type I, classic phenylketonuria or")
2) Phenylalanine ② Hyperphenylalaninemias : defects in phenylalanine hydroxylase (Type I, classic phenylketonuria or PKU) , or in dihydrobiopterin reductase (Types II and III), or in dihydrobiopterin biosynthesis (types IV and V) ④ Diagnoses : * Prenatal DNA probes (Phe hydroxylase or dihydrobiopterin reductase), * Neonatal Fe. Cl 3 (detect urinary phenylpyruvate) ⑤ A diet low in Phe prevent mental retardation of PKU (freq. 1: 10, 000 births) * 효소의 결핍, 진단, 페닐케톤뇨증의 치료 • 페닐케톤뇨증(PKU): - 페닐알라닌 하이드록실레이스의 결핍으로 인해 발병하는 질환. 고페닐 알라닌혈증은 마찬가지로 하이드록실레이스의 보효소인 4중 수소바이 오테린을 합성하거나 분해하는 효소의 결핍으로 인하여 생기는 질환. - 치료하지 않은 PKU 환자의 경우 정신지체, 걸음 혹은 대화장애, 간질, 과운동성, 떨림, 성장장애가 발생. - 신생아가 단백질을 섭취하기 시작한 후 48시간에 채취한 혈액을 이용하 여 조기에 진단. - 치료는 페닐알라닌의 섭취 제한.
 Lysine ① Lys crotonyl-Co. A (Fig. 29 -15) acetyl-Co. A + CO 2")
3) Lysine ① Lys crotonyl-Co. A (Fig. 29 -15) acetyl-Co. A + CO 2 by the reactions of fatty acid catabolism (Fig. 22 -3) ② Hyperlysinemia: elevated lysine (ornithine) inhibits liver arginase hyperammonemia Mitochondria Cytosol Figure 29 -15 Reactions and intermediates in the catabolism of L-lysine
 Tryptophan ① Trp via kynurenine-anthranilate pathway (Fig. 29 -16) N- L-formylkynurenine (by Trp")
4) Tryptophan ① Trp via kynurenine-anthranilate pathway (Fig. 29 -16) N- L-formylkynurenine (by Trp oxygenase + O 2) L-kynurenine ② Kynureninase require PLP and excretion of xanthurenate (Fig. 29 -17) diagnosis of vitamin B 6 deficiency ③ Hartnup disease impaired intestinal & renal transport of Trp ④ Pellagra-like signals and symptoms : Trp deficiency Trp oxygenase (Fig. 29 -17) Figure 29 -16 Catabolism of L-tryptophan
 Methionine ① Met + ATP S-adenosylmethionine (SAM), “active Met” (Fig. 29 -18) Figure")
5) Methionine ① Met + ATP S-adenosylmethionine (SAM), “active Met” (Fig. 29 -18) Figure 29 -18 Formation of S-adenosylmethionine
 Methionine ① Met + ATP S-adenosylmethionine (SAM), “active Met” (Fig. 29 -18) ②")
5) Methionine ① Met + ATP S-adenosylmethionine (SAM), “active Met” (Fig. 29 -18) ② Met SAM S-adenosyl-L-homocysteine-> L-homocysteine -> Cystathione -> L-cysteine + α-ketoglutarate-> propionyl-Co. A (Fig 29 -19) and ultimately succinyl-Co. A (Fig. 20 -2) Intermediates of Citric acid cycle Succinyl. Co. A B 12 coenzyme methyl. Malonyl. Co. A Figure 29 -19 Conversion of methionine to propionyl-Co. A (Fig. 20 -2) metabolism of propionate
 from Met (nutritionally essential AA) : Met SAM homocysteine")
Cysteine Cys (nutritionally nonessential AA) from Met (nutritionally essential AA) : Met SAM homocysteine (Fig. 29 -19) L-Homecysteine + Ser Cystathionine L-Cys and homoserine (Fig. 27 -9) SAM (Fig. 29 -19) Propionyl-Co. A Figure 27 -9 Conversion of homocysteine and serine to homoserine and cysteine

The Initial Reactions are Common to All Three Branched-chain Amino Acids : first three reactions, analogous to fatty acid catabolism (Fig. 22 -3) Transamination , Oxidative decarboxylation (decarboxylase, a transacylase, and a dihydrolipoly dehyrogenase resembles pyruvate dehydrogenase), Dehydrogenation Maple syrup urine disease (branched-chain ketonuria) Figure 29 -20 The analogous first three reactions in the catabolism of leucine, valine, and isoleucine


The Initial Reactions are Common to All Three Branched-chain Amino Acids * Subsequent catabolism of each AAs (Fig. 29 -21, 22, 23) (Fig. 29 -21) Acetoacetate + Acetyl- Co. A (Fig. 29 -23) Succunyl-Co. A (Fig. 29 -22) Propionyl-Co. A + Acetyl-Co. A

Chapter 30. Conversion of Amino Acids to Specialized Products Biomedical importance ① Modified amino acid in protein for a specific function ② Amino acid derivatives: heme, purines, pyrimidines, hormones, neurotransmitters, and biological active peptides ③ Small peptides or peptide like molecules: histidine ④ Neurotransmitters and many drugs

α-amino acids Alanine: - Carrier of ammonia and of the carbons of pyruvate from skeletal muscle to liver via cori cycle (Fig. 20 -4) glycogen lactate Figure 28 -3 The glucose-alanine cycle Fig 20 -4 Cori cycle glycogen Figure 28 -4 Summary of amino acid exchange between organs immediately after feeding
 Figure 30")
α-amino acids Arginine: - formamidine donor for creatine synthesis (Fig. 30 -12) Figure 30 -12 Biosynthesis of creatine Arg Figure 30 -1 Arginine, ornithine, and proline metabolsm
 - Ornithine")
α-amino acids Arginine: - formamidine donor for creatine synthesis (Fig. 30 -12) - Ornithine carbons of Polyamines (putrescine, spermine and spermidine) (Fig. 30 -8) Figure 30 -1 Arginine, ornithine, and proline metabolsm

Figure 30 -8 Intermediates and enzymes that participate in the biosynthesis of spermidine and spermine

Spermidine Methionine NH 3 Ornithine SAM Spermine Figure 30 -8 Intermediates and enzymes that participate in the biosynthesis of spermidine and spermine NH 3

Polyamine oxidase Figure 30 -9 Catabolism of polyamines

α-amino acids Arginine: - formamidine donor for creatine synthesis - Ornithine Polyamine (putrescine spermine and spermidine )(Fig. 30 -8) - Arg NO (neurotransmitter, smooth muscle relaxant, vasodialtor) + citrulline by NOS Figure 30 -1 Arginine, ornithine, and proline metabolsm
 by forming 4")
α-amino acids Cysteine: - biosynthesis of coenzyme A (Fig. 44 -18) by forming 4 -phosphopanthothenoyl-cysteine (Fig. 30 -2) - taurine conjugates with bile acids (Fig. 26 -7; Biosynthesis and degradation of bile acids) Figure 30 -2 The reaction catalyzed by phosphopantothenate-cysteine ligase Figure 30 -3 Conversion of cysteine to taurine

α-amino acids Glycine: ① Metabolites and pharmaceuticals excreted as water-soluble glycine conjugates: glycocholic acid (Chap 26) and hippuric acid (Fig. 30 -4) formed from the food additive bezoate Figure 30 -4 Biosynthesis of hippurate Glycine

α-amino acids Glycine: ① Metabolites and pharmaceuticals excreted as water-soluble glycine conjugates: glycocholic acid (Chap 26), hippuric acid (Fig. 30 -4) ② Gly incorporated into creatine (Fig. 30 -12) Glycine Arg SAM Creatine Figure 30 -12 Biosynthesis of creatine and creatinine

α-amino acids Glycine: ① Metabolites and pharmaceuticals excreted as water-soluble glycine conjugates glycocholic acid (Chap 26), hippuric acid (Fig. 30 -4) ② Gly incorporated into creatine (Fig. 30 -12) ③ Heme (Chap. 31) - Nitrogen and -Carbon are incorporated into pyrrole rings and methylene bridge carbons of heme ④ Purine biosynthesis (Fig. 33 -1) - Entire glycine molecule becomes atoms of 4, 5, and 7 of purines Glycine

α-amino acids Histidine ① Decarboxylation of His to histamine by broad specificity of aromatic L-amino acid decarboxylase (Dopa, 5 -hydroxytryptophan, Phe, Tyr, and Trp) (Fig. 30 -5) : 알레르기 반응, 위산분비, 모든 조직에 존재. ② Histidine compounds present in the human body ergothioneine, carnosine, and anserine (Fig. 30 -6) - Unknown functions, - major constituents of excitable tissues, brain, and skeletal muscle ③ Wilson’s disease low urinary levels of 3 -methyl. Histidine Histamine Figure 30 -5 The reaction catalyzed by histidine decarboxylase Figure 30 -6 Derivatives of histidine
 - major nonprotein")
α-amino acids Methionine ① S-Adenosylmethione : methyl donor (Fig. 30 -7) - major nonprotein fate - principal source of methyl group in the body Methionine (Figure 29 -19) SAM Figure 30 -7 Biosynthesis of S-adenosylmethionine, catalyzed by methionine adenosyltransferase (MAT)
 - major nonprotein")
α-amino acids Methionine ① S-Adenosylmethione : methyl donor (Fig. 30 -7) - major nonprotein fate - principal source of methyl group in the body ② Biosynthesis of polyamine: spermine and spermidine (Fig. 31 -4) Spermidine and spemine : growth factors (Fig. 30 -8) Catabolism of polyamines (Fig. 30 -9) Multiple positive charge : association with DNA and RNA

Spermidine Methionine NH 3 Ornithine SAM Spermine Figure 30 -8 Intermediates and enzymes that participate in the biosynthesis of spermidine and spermine NH 3

Polyamine oxidase Figure 30 -9 Catabolism of polyamines
 (Chap. 24) • Biosynthesis of purine")
α-amino acids Serine • Biosynthesis of sphingosine (ceramide) (Chap. 24) • Biosynthesis of purine , pyrimidines (Chap. 33) - carbons 2 and 8 of purines - methyl group of thymine • Conversion of serine and homocysteine : cystathione β-synthase : Cysteine Serine Met SAM se a h t t s cy ni o i h t a β e n n y s - Figure 27 -9 Conversion of homocysteine and serine to homoserine and cysteine Cysteine
 Trp 5 -hydroxy. Trp by Trp hydroxylase serotonin (5 -hydroxytryptamine)")
α-amino acids Tryptophan (Trp) Trp 5 -hydroxy. Trp by Trp hydroxylase serotonin (5 -hydroxytryptamine) by decarboxylation (Fig. 30 -10) * serotonin : a potent vasoconstrictoor, stimulator of smooth muscle contraction Tryptophan hydroxylase 5 -OH Tryptophan Serotonin Figure 30 -10 Biosynthesis and metabolism of serotonin and melatonin

MAO inhibitor MAO Serotonin N-acetylation of serotonin and O-methylation in pineal body Melatonin Figure 30 -10 Biosynthesis and metabolism of serotonin and melatonin
 * In neural cells, Tyr (L-dopa , dopamine) norepinephrine and")
α-amino acids Tyrosine (Catecholamines) * In neural cells, Tyr (L-dopa , dopamine) norepinephrine and epinephrine (Fig. 30 -11) * In melanocytes, different enzyme hydroxylase Tyr (tyrosinase melanin) * In adrenal medulla, phenylethanolamine-N-methyltransferase utilizes SAM epinephrine * Tyr is also precursor of triiodothyronine and thyoxine (Ch. 42) Norepinephrine Tyrosine Dopamine SAM Epinephrine Dopa Figure 30 -11 Conversion of tyrosine to epinephrine and norepinephrine in neuronal and adrenal cells
 ② 24 -hour urinary")
Creatinine ① Creatinine in muscle from creatine (Fig. 30 -12) ② 24 -hour urinary excretion of creatinine is proportionate to muscle mass ③ Gly, Arg, and Met creatinine biosynthesis Glycine Arg Methionine SAM Creatine Creatinine Figure 30 -12 Biosynthesis of creatine and creatinine
")
Non-α-amino acids - Present in tissues in a free form -alanine, -aminoisobutyrate, γ-Aminobutyrate (GABA) -alanyl dipeptides : Activate myosin ATP ase ; Chelate copper ; Enhance copper uptake
 as inhibitory neurotransmitter ② Glu GABA by L-Glu")
Non-α-amino acids γ-Aminobutyrate ① γ-Aminobutyrate (GABA) as inhibitory neurotransmitter ② Glu GABA by L-Glu decarboxylase (Fig. 30 -13) ③ Transamination of GABA succinate semialdehyde hydroxybutyrate (reduction) or succinate (oxidation) Glutamate GABA Hydroxybutyrate Transamination NH 4+ Succinate semialdehyde Figure 30 -13 Metabolism of g-aminobutyrate Succinate
- Slides: 67